
Age-associated memory decline greatly affects the quality of life of a large proportion of older individuals1. Neuronal engrams in the hippocampus are critically involved in the formation, storage and recall of memories7. The ability of the hippocampus to store new information in neuronal engrams declines with age8,9, but the mechanisms underlying memory loss are incompletely understood. The gastrointestinal microbiome has recently emerged as an important factor in the regulation of cognition and has been implicated as a modifiable peripheral signal that may contribute to age-associated memory loss4,5,6,10,11,12,13. However, the circuits by which gut microbial signals are transmitted to the brain to modulate memory remain largely unclear. Here we chart a microbiome–gut–brain pathway spanning intestinal metabolites, innate immune responses, vagal signalling and hippocampal memory encoding that impacts the rate of cognitive decline. Our results provide several targets for peripheral intervention and suggest that inflammation-induced interoceptive dysfunction might be a generalizable principle underlying age-associated cognitive decline.
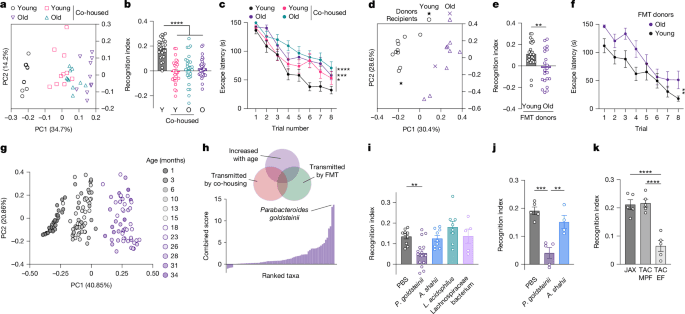
To investigate the consequences of age-associated microbiome alterations on cognitive ageing, we experimentally uncoupled the microbiome age from the host age by transferring the gut microbial community from aged mice to young recipients. We achieved accelerated microbiome ageing in young mice by co-housing 2-month-old with 18-month-old mice, leading to an equilibration of the microbial communities between young and old mice that resembled an old-like state (Fig. 1a). Physical health, as assessed by frailty scoring, was unaltered by microbiome equilibration (Extended Data Fig. 1a).
a–c, Principal coordinates analysis (PCoA) plot of microbiome dissimilarity (a), NOR (b) and Barnes maze escape latency (c) in young (Y, 2 months) and old (O, 18 months) mice after one month of co-housing. d–f, PCoA plot of microbiome dissimilarity (d), NOR (e) and Barnes maze escape latency (f) in germ-free mice one month after faecal microbiota transfer (FMT) from young and old donors. g, PCoA plot of metagenomic taxonomy in wild-type mice across the lifespan. h, Identification of bacterial species increased in ageing and transferred by co-housing as well as FMT. i–k, NOR in germ-free (i) or antibiotic-treated (j) mice colonized with the indicated species, or in mice with natural variability in P. goldsteinii abundance (k). EF, excluded flora; JAX, Jackson Laboratory; L. acidophilus, Lactobacillus acidophilus; MPF, murine pathogen free; PBS, phosphate-buffered saline; TAC, Taconic. Error bars indicate mean ± s.e.m. **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 2.
Notably, one month of co-housing with aged mice impaired the short-term memory of young mice in the novel object recognition (NOR) task (Fig. 1b), which was maintained during prolonged co-housing (Extended Data Fig. 1b,c). This phenotype was observed in both female (Fig. 1b) and male (Extended Data Fig. 1d) mice, and in various combinations of young and aged mice obtained from different vendors (Extended Data Fig. 1e–g); explorative behaviour, however, was not affected by co-housing (Extended Data Fig. 1h–j). The impact of the aged microbiome on learning and memory was likewise observed in the Barnes maze assay—a long-term spatial learning and memory task (Fig. 1c and Extended Data Fig. 1k).
We next sought to disentangle the impact of co-housing on intestinal microbial community composition from its social effects. To this end, we pursued four orthogonal experimental strategies. First, we co-housed young mice with different young mice, which did not affect their cognitive behaviour (Extended Data Fig. 1l). Second, we colonized young germ-free mice with faecal microbiome samples obtained from either young or aged donors. Microbiome transplantation recapitulated the composition of donor microbiomes in the recipients (Fig. 1d), thus establishing age-associated microbiome alterations in young mice without co-housing. As in the co-housing setting, accelerating microbiome ageing in young mice abolished learning and memory performance in the NOR and Barnes maze tasks (Fig. 1e,f and Extended Data Fig. 1m) without altering explorative behaviour (Extended Data Fig. 1n). Our third experimental strategy to explore causality for the microbiome in driving age-associated cognitive decline was to perform co-housing experiments under germ-free conditions. We aged germ-free mice to over 18 months and co-housed them with young germ-free mice in sterile isolators. Notably, young germ-free mice retained their full cognitive capacity despite co-housing (Extended Data Fig. 1o), indicating that the social effect of co-habitation with aged animals was not sufficient to induce the cognitive deficit. When we aged germ-free mice for these experiments, we also noticed that their rate of cognitive decline was delayed compared with conventional mice, still showing normal learning and memory at 18 months (Extended Data Fig. 1p). Fourth, we used several antibiotic strategies to ablate microbial colonization before, during or after co-housing of young and old mice (Extended Data Fig. 1q). Importantly, co-housing of young mice with aged mice whose microbiome had been ablated with broad-spectrum antibiotics before co-housing did not result in impaired NOR (Extended Data Fig. 1r). Furthermore, antibiotics administered concurrently to co-housing also protected young mice from the detrimental impact of co-housing (Extended Data Fig. 1r), thus recapitulating the results obtained from co-housing under germ-free conditions. We also sought to determine whether the negative impact of microbiome ageing was reversible. To this end, we treated young mice with broad-spectrum antibiotics after they developed the co-housing-induced cognitive deficit. Not only did two weeks of antibiotic treatment restore memory performance in co-housed young mice, but even aged mice showed an improvement in cognitive ability after antibiotic treatment (Extended Data Fig. 1r,s). Collectively, these findings suggest that at advanced age, microbiome elements contribute to cognitive decline—a trait that is transmissible to young mice and preventable by antibiotic treatment.
We next sought to use these experimental microbiome perturbations to identify elements of the bacterial microbiota that are involved in the regulation of cognitive decline. We identified candidate taxa using two criteria: (1) their relative abundance increases with age, and (2) they are transmitted to young animals by co-housing and microbiome transplantation. To characterize microbiome changes over the entire lifespan, we followed a cohort of 15 male C57BL/6 mice, longitudinally assessed health, and performed metagenomic sequencing as well as proteomics on faecal content every 2–4 months from weaning to death (Extended Data Fig. 1t). The cohort showed an onset of frailty and mortality at the age of two years, a mean lifespan of 955 days and a maximal lifespan of 1,210 days (Extended Data Fig. 1u,v). We assigned taxonomic identities to metagenomic reads using Kraken 2 (ref. 14). Age was a major driver of variability in the taxonomic composition of the microbiota (Fig. 1g and Extended Data Fig. 2a), and 1,133 species showed significant alterations in relative abundance over the lifespan (Extended Data Fig. 2b). The acquisition of age-associated bacterial community changes was associated with alterations in the metagenomic coding capacity of the microbiome, as determined by the change in relative abundance of KEGG orthologues over time (Extended Data Fig. 2c–e).
We then ranked taxonomic elements according to our criteria for identifying possible drivers of cognitive decline (Fig. 1h and Supplementary Table 1). The top candidate was Parabacteroides goldsteinii, whose abundance increased with age and was transmissible by co-housing and microbiota transplantation (Fig. 1h and Extended Data Fig. 2f–h). Stool proteomics confirmed an age-associated increase in peptides from Parabacteroides (Extended Data Fig. 2i). Notably, colonization of germ-free or antibiotic-treated mice with P. goldsteinii induced cognitive impairment (Fig. 1i,j and Extended Data Fig. 2j,k). Furthermore, young mice from a facility with naturally high levels of P. goldsteinii15 showed reduced memory function (Fig. 1k and Extended Data Fig. 2l). Other bacteria, such as Alistipes and Lachnospiraceae, whose abundance likewise changed with age and was transmissible by co-housing (Extended Data Fig. 2m,n), or Lactobacillus, which did not show any changes with age (Extended Data Fig. 2o), did not impact cognitive function (Fig. 1i,j and Extended Data Fig. 2j,k). Collectively, these findings suggest that certain microbiome alterations over the lifespan—including the outgrowth of P. goldsteinii—influence the rate of cognitive decline in mice.
Learning and memory are highly dependent on the hippocampus16,17. We thus explored whether microbiome ageing and P. goldsteinii outgrowth impacted hippocampal function. Adult hippocampal neurogenesis declines with age and has been linked with learning and memory capacity18,19. We noted a strong reduction in hippocampal neurogenesis in old mice, but co-housed young mice were not affected (Extended Data Fig. 3a,b). Similarly, enhanced inflammation in the hippocampus, as indicated by astrogliosis, was observed in aged mice but not transmissible to young mice by co-housing (Extended Data Fig. 3c,d). We also did not notice any differences in structural plasticity of dendritic spines (Extended Data Fig. 3e,f). These results indicate that hippocampal neurogenesis, inflammation or dendritic spine morphology are unlikely to account for the microbiome impact on learning and memory.
We thus used an unbiased RNA-sequencing-based approach to assess hippocampal molecular changes in mice with different host and microbiome ages. We classified differentially expressed genes into four categories according to their correlation with age or cognitive performance of the host (Fig. 2a). Notably, immediate-early gene activation in response to novel object exposure was robustly observed in young mice but blunted in both aged groups as well as young mice co-housed with old mice (Fig. 2a,b and Extended Data Fig. 3g). As immediate-early gene expression is an indicator of neuronal activation, we next explored whether specific areas of the hippocampus contributed to this reduction of neuronal responses. Staining for FOS revealed that CA3, CA1 and dentate gyrus of co-housed young mice showed impaired activation in response to novelty exposure (Fig. 2c,d). Furthermore, germ-free recipients of microbiota samples from young mice showed a strong increase in dentate gyrus activity in response to a novel object, but FOS responses were blunted in recipients of microbiota from old donors (Fig. 2e,f). The rescue of cognitive abilities in aged antibiotic-treated or germ-free mice was associated with restoration of hippocampal FOS responses to a novel object (Extended Data Fig. 3h,i). Conversely, colonization of young mice with the age-associated bacterial species P. goldsteinii inhibited hippocampal responses to novel object exposure (Fig. 2g,h). These experiments suggest that age-associated microbiome changes impact memory function by altering neuronal responses in the hippocampus.
a, Heatmap of differentially expressed hippocampal genes in singly-housed and co-housed young and aged mice. b, Expression of hippocampal immediate-early genes. c–h, Representative images (c,e,g) and quantifications (d,f,h) of FOS+ neurons in the hippocampus of co-housed mice (c,d), germ-free recipients of microbiomes from young and old donors (e,f) and P. goldsteinii-colonized mice (g,h). Error bars indicate mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, 100 μm. Exact n and P values are presented in Supplementary Table 2.
Of note, the age-associated changes in FOS responses were not unique to the hippocampus. Several additional brain areas—including the nucleus tractus solitarii (NTS)6, and somatosensory and entorhinal cortices—showed reduced neuronal activation in both old and co-housed young mice (Fig. 3a,b and Extended Data Fig. 4). Given the role of these areas in sensory processing, we postulated that ageing leads to reduced transduction of interoceptive information through gut-innervating sensory neurons. We took advantage of the fact that a large fraction of both vagal and spinal afferents expresses the vanilloid receptor TRPV1 (Extended Data Fig. 5a) and assessed the cognitive performance of Trpv1DTA mice, which are devoid of TRPV1-expressing neurons. Notably, young Trpv1DTA mice showed reduced hippocampal activation and phenocopied old mice in the NOR task (Fig. 3c–e) while retaining normal gross hippocampal architecture (Extended Data Fig. 5b). The same effect was observed after chemogenetic silencing of TRPV1+ neurons (Fig. 3f–h and Extended Data Fig. 5c,d). We thus speculated that inhibition of sensory neurons may underlie the negative impact of age-associated microbiome changes on hippocampal function. To test this, we chemogenetically activated TRPV1+ neurons in young mice co-housed with aged mice6. Remarkably, activation of sensory neurons fully restored their hippocampal FOS activity and cognitive performance, while leaving the NOR ability of young controls unaltered (Fig. 3i–k). A similar effect was observed with low doses of the TRPV1 agonist capsaicin, which rescued NTS activation, hippocampal responses and memory function in aged mice and co-housed young mice (Fig. 3l–p and Extended Data Fig. 5e–g). Capsaicin treatment was likewise able to overcome the detrimental effect of P. goldsteinii on hippocampal memory (Fig. 3q). Expectedly, the beneficial effect of capsaicin on cognition was dependent on the presence of TRPV1+ neurons (Extended Data Fig. 5h).
a,b, FOS responses in the NTS of singly-housed and co-housed young and aged mice. c–t, Representative images of FOS+ hippocampal neurons (c,f,i,l,r), FOS quantifications (d,g,j,m,o,s) and NOR (e,h,k,n,p,q,t) in Trpv1DTA mice (c–e); Trpv1AAV-hM4Di mice (f–h); co-housed Trpv1AAV-hM3Dq mice (i–k); capsaicin-treated young and old mice (l–n); capsaicin-treated co-housed young and old mice (o,p); P. goldsteinii-colonized mice (q); and Trpv1-Cre/Phox2b-FlpOhM4Di mice (r–t). u–y, Schematic of in vivo calcium imaging of nodose ganglia in response to duodenal Ensure infusion (u) and heatmaps (v), quantification of responsive neurons (w), z-scores over time (x) and mean responses (y) of vagal neurons in young mice and young mice co-housed with aged mice. Arrow indicates start of Ensure infusion. CNO, clozapine-N-oxide. Error bars indicate mean ± s.e.m. Scale bars, 100 μm. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 2. Image in u created in BioRender; T, C. https://biorender.com/s2mmfpa (2026).
Given that TRPV1 is broadly expressed by both spinal and vagal afferents, as well as certain areas of the brain, we sought to more precisely define the population of neurons that mediate the microbial impact on memory function, using marker-guided targeting of sensory subpopulations20,21. We first silenced neurons expressing Phox2b, which marks vagal but not spinal afferents (Extended Data Fig. 5a). Chemogenetic inhibition of Phox2b-expressing neurons in young mice was sufficient to recapitulate memory deficits and hippocampal FOS suppression (Extended Data Fig. 5i–l). We then used intersectional genetic targeting of PHOX2B+ TRPV1+ neurons to more selectively target vagal Trpv1-expressing cells—as confirmed by whole-animal imaging (Extended Data Fig. 5m)—which likewise abrogated hippocampal novelty responses and object memory (Fig. 3r–t). By contrast, targeting of spinal Trpv1-expressing afferents—through intrathecal injection of resiniferatoxin or surgical ablation of gut-innervating spinal neurons through coeliac ganglionectomy—did not impact cognitive performance (Extended Data Fig. 6a–c). These findings suggest that impaired vagal function might be involved in the intestinal regulation of age-associated cognitive decline. To directly address this hypothesis, we performed calcium imaging in response to intestinal nutrient infusion of individual vagal neurons in young mice and young mice co-housed with aged mice (Fig. 3u). Notably, nodose ganglia from young mice harbouring an age-associated microbiome showed diminished vagal activation, as evidenced by a reduced number of responding neurons and a lower magnitude of neuronal activation (Fig. 3v–y).
Vagal PHOX2B+ TRPV1+ neurons are composed of several neuronal subsets22,23, including gut-innervating neurons expressing the cholecystokinin A receptor (Cckar) (Extended Data Fig. 5a). We found that ablation of CCKAR+ neurons by nodose injection of the cholecystokinin-conjugated neurotoxin saporin24,25,26 (CCK-SAP; Extended Data Fig. 6d) impaired cognitive performance (Extended Data Fig. 6e), in line with past observations in rats26. As CCKAR+ neurons respond to intestinal CCK peptide27 and transmit information between the gut and hippocampus28,29, we sought to determine whether CCK stimulation of vagal afferents is sufficient to restore cognitive performance in aged mice. Notably, after peripheral administration of CCK, the cognitive performance of aged mice and co-housed young mice was indistinguishable from that of control young mice, and hippocampal FOS responses were restored (Extended Data Fig. 6f–k). As CCK not only acts as a gastrointestinal vagal activator but also exerts hormonal effects on other tissues30, we determined whether its memory-enhancing effect depended on sensory neurons. Denervated Trpv1DTA mice were unresponsive to CCK administration (Extended Data Fig. 6l), suggesting that sensory neurons are necessary for the effect of CCK on hippocampal function. The same effect was achieved with glucagon-like peptide 1 (GLP1)—another intestinal peptide that stimulates the vagus nerve (Extended Data Fig. 6m,n). Consistently, the GLP1 receptor agonist liraglutide enhanced memory function in aged mice (Extended Data Fig. 6o). Although these gut peptides were able to restore cognitive function, their endogenous levels were unchanged during ageing and P. goldsteinii colonization (Extended Data Fig. 6p–s). Furthermore, the CCKAR inhibitors devazepide and A-65186 did not induce cognitive deficits (Extended Data Fig. 6t,u), suggesting that reduced gut hormone levels do not account for the age-associated interoceptive dysfunction that drives cognitive decline.
We thus sought to identify the mechanisms by which age-associated microbiome alterations impact vagal signalling and memory function. Microbial interventions that induced cognitive dysfunction were not associated with enhanced intestinal barrier permeability (Extended Data Fig. 7a). We therefore tested whether their activity was mediated by a secreted molecule. To this end, we cultured P. goldsteinii and administered culture supernatants to mice through oral supplementation (Fig. 4a). Notably, the size-filtered supernatant fraction was sufficient to induce cognitive decline in recipient mice (Fig. 4b). By contrast, supernatants from Alistipes shahii cultures did not show activity (Fig. 4b). The activity of P. goldsteinii supernatants was retained after 3 kDa size filtration and observed across different culture media (Fig. 4a and Extended Data Fig. 7b,c). We thus concluded that the effect of P. goldsteinii on hippocampal function was probably mediated by small metabolites. To identify candidate molecules, we performed untargeted metabolomics on the culture supernatants (Fig. 4c). Among the molecules most strongly enriched in P. goldsteinii cultures was the medium-chain fatty acid (MCFA) 3-hydroxyoctanoic acid (3-HOA). Remarkably, oral supplementation of this metabolite, which increased its concentrations both locally and systemically but not in the brain (Extended Data Fig. 7d), was sufficient to recapitulate reduced hippocampal responses and impaired object recognition (Fig. 4d–f), suggesting that it may have an important role in the modulation of age-associated cognitive decline. To determine whether this function was unique to 3-HOA or generalizable to other MCFAs, we examined decanoic and dodecanoic acids, both of which had not been detected in our initial metabolomics experiment due to their water-insolubility. These MCFAs were likewise increased by colonization with P. goldsteinii and had a comparable effect on cognition following oral administration (Fig. 4g and Extended Data Fig. 7e–g). Similar to an aged microbiome, MCFA treatment blunted vagus nerve activation, reduced neuronal stimulation in the NTS, and impaired hippocampal responses to novel object exposure (Fig. 4h–o).
a, Schematic of workflow. b, NOR in mice receiving filtered supernatants from P. goldsteinii and A. shahii cultures via oral gavage. c, Volcano plot of metabolites from P. goldsteinii and A. shahii cultures. d–g, Representative images of FOS+ dentate gyrus neurons (d), FOS quantifications (e) and NOR (f,g) in mice treated with 3-HOA (d–f), or with decanoic or dodecanoic acid (g). h–k, In vivo calcium imaging of nodose ganglia in response to duodenal Ensure infusion of mice treated with decanoic acid, shown as heatmaps (h), z-scores over time (i), mean responses (j) and peak responses (k). Arrow indicates start of Ensure infusion. l,m, Representative images (l) and quantification of FOS+ neurons (m) in the NTS of decanoic acid-treated mice and controls. n–y, Representative images of FOS+ dentate gyrus neurons (n,p,s,v), FOS quantifications (o,q,t,w) and NOR (r,u,x,y) in: wild-type (n,o) or Gpr84−/− mice treated with decanoic acid (p–r); mice given embelin, 3-HOA, PBI-4050 or capsaicin (s–w); P. goldsteinii-colonized mice treated with PBI-4050 (x); or old mice treated with PBI-4050 (y). Error bars indicate mean ± s.e.m. Scale bars, 100 μm. *P < 0.05, **P < 0.01, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 2. Images in a created in BioRender; T, C. https://biorender.com/p6614lw (2026).
MCFAs can be found as constituents of the lipid A component in bacterial lipopolysaccharide (LPS)31. We therefore speculated that microbiota changes may cause elevated MCFA levels during ageing. Consistently, we found that the abundance of MCFAs in the intestinal lumen increased with age in conventionally colonized, but not germ-free or antibiotic-treated mice (Extended Data Fig. 7h–k). Furthermore, elevated luminal MCFA levels were transmissible from old to young mice by co-housing (Extended Data Fig. 7l). We also hypothesized that interventions aimed at reducing bacterial MCFA production could possibly decelerate the rate of cognitive decline. Given the potential of bacteriophages (phages) as modulators of the intestinal microbial community, we screened phages for their ability to restore memory function in aged mice. We consistently observed cognitive improvement in aged mice treated with φPDS1 (ref. 32)—a phage targeting Parabacteroides distasonis (Extended Data Fig. 7m,n). Other phages, such as φAPCM01, a Streptococcus mutans-targeting virus that shares its class (Caudoviricetes) and morphology (siphovirus) with φPDS1, or φX174, a taxonomically distinct phage targeting Escherichia coli, did not affect memory function (Extended Data Fig. 7m,n). Although P. distasonis was absent from mice used in our experiments, we speculated that φPDS1 may reduce the abundance of its taxonomic relative P. goldsteinii; however, each phage showed strong host specificity, and P. goldsteinii levels were not impacted by φPDS1 both in vitro and in vivo (Extended Data Fig. 7o–r). In addition to lysis, phage infection can influence the biology of the bacterial host through transcriptional alterations—often provoking changes in cell surface structures33,34. To probe the transcriptional response of P. goldsteinii to φPDS1, we performed RNA sequencing of phage-infected cultures and compared them with cultures exposed to φAPCM01. We indeed noted strong transcriptional differences between the two conditions, with many differentially expressed genes affecting outer cell wall biology (Extended Data Fig. 7s–u). Importantly, φPDS1 reduced the levels of MCFAs detected in the intestinal lumen of aged mice (Extended Data Fig. 7v,w). Together, these findings suggest that phage targeting of intestinal bacteria may reduce intestinal MCFA levels and alleviate cognitive decline.
We next wanted to better understand how MCFAs can influence cognitive function. Several MCFAs are highly potent agonists of the surface receptor GPR84 (ref. 35). We therefore investigated whether the impact of MCFAs on memory was mediated by GPR84 signalling. The first evidence came from the observation that DBA mice—which harbour a point mutation36 that renders them naturally deficient in GPR84 (Extended Data Fig. 8a)—were resistant to the impact of 3-HOA on cognitive function, and showed delayed onset of cognitive decline compared with C57BL/6 mice despite being colonized by P. goldsteinii (Extended Data Fig. 8b–d). GPR84-deficient C57BL/6 mice were likewise protected from the impact of MCFAs on hippocampal responses and memory dysfunction (Fig. 4p–r). Furthermore, the GPR84 agonist embelin phenocopied the impact of MCFAs on hippocampal FOS responses and NOR (Fig. 4s,t), while the GPR84 inhibitor PBI-4050 negated the effect of 3-HOA on cognitive performance, similar to the vagal stimulant capsaicin (Fig. 4u). Furthermore, PBI-4050 restored neuronal activation in the hippocampus, counteracted the detrimental impact of P. goldsteinii on memory function, and reestablished youthful NOR in aged mice (Fig. 4v–y). Collectively, these results indicate that the impact of MCFAs on memory formation is mediated by GPR84, and that GPR84 inhibition may provide protection from age-associated cognitive decline.
The expression of GPR84 is largely confined to myeloid cells37. Indeed, in a single-cell survey of intestinal immune cells38, we found that Gpr84 was only expressed by macrophages, monocytes and neutrophils (Fig. 5a and Extended Data Fig. 8e,f). We thus tested a possible involvement of myeloid cells in microbiome- and MCFA-induced cognitive ageing. Of note, the CSF1R inhibitor PLX-3397, which depletes myeloid cells, restored cognitive function both in the co-housing setting and after 3-HOA administration (Fig. 5b,c). As CSF1R inhibition ablates myeloid cells both centrally and peripherally39 (Extended Data Fig. 8g,h), we sought to use orthogonal strategies to target peripheral macrophages while sparing microglia in the brain. Importantly, clodronate liposomes, which deplete phagocytic cells but do not cross the blood–brain barrier (Extended Data Fig. 8i,j), likewise rescued the detrimental effect of 3-HOA on NOR (Fig. 5d). Similarly, mice with a genetic deficiency in CCR2, which leads to impaired recruitment of myeloid cells to peripheral tissues but not the brain40 (Extended Data Fig. 8k–n), were equally resistant to the effect of 3-HOA (Fig. 5e). Myeloid cell ablation improved the response of vagal neurons to nutrient stimulation (Fig. 5f–h), and hippocampal responses to novelty exposure (Fig. 5i,j).
a, Uniform manifold approximation and projection (UMAP) of single-cell transcriptomes of colonic CD45+ cells highlighting Gpr84 expression. b,c, NOR in co-housed (b) and 3-HOA-treated (c) mice receiving PLX3397. d,e, NOR in 3-HOA-treated mice receiving clodronate liposomes (d) or with Ccr2 deficiency (e). f–h, In vivo calcium imaging of nodose ganglia in response to duodenal Ensure infusion of decanoic acid-treated mice receiving PLX3397, shown as heatmaps (f), z-scores over time (g) and peak responses (h). Arrow indicates start of Ensure infusion. i,j, Representative images of FOS+ dentate gyrus neurons (i), and FOS quantification (j) in 3-HOA-treated mice receiving clodronate liposomes or with Ccr2 deficiency. KO, knockout. k–n, Schematic (k,m) and NOR (l,n) of decanoic-acid-treated wild-type (WT) mice harbouring bone marrow transplants from Gpr84−/− (k,l) or 50/50 mixed Gpr84−/−/Ccr2−/− mice (m,n). o,p, NOR in co-housed (o) and 3-HOA-treated (p) mice receiving anti-TNF antibody. q, NOR in co-housed mice receiving anti-IL-1β antibody. r, NOR in Nlrp3−/− mice treated with 3-HOA. s, NOR in Phox2bCre-Il1r1fl/fl mice treated with decanoic acid. t, NOR in Trpv1AAV-hM3Dq mice treated with IL-1β and CNO. Error bars indicate mean ± s.e.m. Scale bar, 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 2. Images in k and m created in BioRender; T, C. https://biorender.com/lbleqjs (2026).
To directly test whether the effect of GPR84 was mediated by myeloid cells, we generated bone marrow chimeras, in which we reconstituted irradiated recipient mice with bone marrow from either wild-type or GPR84-deficient mice, verified efficient engraftment and then treated the recipients with MCFA (Fig. 5k and Extended Data Fig. 8o,p). In these mice, cognitive function was only retained when GPR84 was lacking from the haematopoietic compartment (Fig. 5l). To increase specificity for myeloid cells, we used mixed bone marrow chimeras, in which we reconstituted irradiated recipients with a 50:50 mixture of GPR84- and CCR2-deficient bone marrow (Fig. 5m). In these mice, peripheral myeloid cells selectively lack GPR84 expression (Extended Data Fig. 8q), and their cognitive performance was intact despite MCFA treatment (Fig. 5n). The depletion of other immune cell types did not impact performance in the NOR task (Extended Data Fig. 9a–h). These findings highlight an important role for GPR84 on peripheral myeloid cells in driving the impact of MCFAs on cognitive ageing.
GPR84 signalling triggers the production of pro-inflammatory cytokines, such as tumour necrosis factor (TNF) and interleukin-1β (IL-1β) (refs. 41,42; Extended Data Fig. 9i). We noticed that acquisition of an aged microbiome led to increased expression of inflammatory cytokines locally in the gastrointestinal tract as well as in adipose tissue, but not systemic sites nor the brain (Extended Data Fig. 9j–m). We hypothesized that the lipophilic nature of MCFAs may lead to their preferential accumulation in adipose tissue43, and that mesenteric adipose tissue might be a source of inflammatory cytokines in the gastrointestinal tract. Indeed, in mice treated with MCFAs, we observed an increased concentration within adipose tissue, as well as elevated cytokine expression in both mesenteric and inguinal adipose tissue (Extended Data Fig. 10a–c). This was also seen in aged C57BL/6 but not aged DBA/2 mice (Extended Data Fig. 10d–f), indicating that GPR84 might be involved in driving age-associated inflammation in response to MCFA accumulation.
Finally, we sought to determine the functional relevance of peripheral inflammatory cytokines for memory loss. We first confirmed that administration of either TNF or IL-1β was sufficient to impair cognitive function (Extended Data Fig. 10g,h). This effect was rescued by CCK treatment (Extended Data Fig. 10g), indicating that vagal activation was able to restore cognitive performance despite ongoing inflammatory responses. Second, a TNF-neutralizing antibody rescued memory function in aged mice as well as co-housed young mice and protected against the detrimental effect of 3-HOA supplementation (Fig. 5o,p). Similarly, IL-1β neutralization protected aged and co-housed young mice (Fig. 5q). Furthermore, deficiency in NLRP3—a GPR84-induced inflammasome component required for proteolytic processing of IL-1β (refs. 41,44)—likewise conferred resistance against the cognitive impact of 3-HOA treatment (Fig. 5r). We also investigated the cellular targets of TNF and IL-1β that contribute to cognitive decline. Although TNF receptor expression was predominantly required on haematopoietic cells (Extended Data Fig. 10i), deletion of the receptor of IL-1β on PHOX2B-expressing cells counteracted the effect of MCFAs on memory function (Fig. 5s), suggesting that IL-1β signalling on vagal sensory neurons impaired their function. Consistently, chemogenetic reactivation of sensory neurons protected mice from the detrimental effect of IL-1β on memory (Fig. 5t). Altogether, our findings suggest a model whereby ageing leads to changes in the gastrointestinal milieu, including the outgrowth of P. goldsteinii and accumulation of MCFAs. These metabolites, in turn, drive pro-inflammatory myeloid cell responses through GPR84 signalling, thereby impairing vagal activity, hippocampal responses and memory function (Extended Data Fig. 10j).
Memory loss is one of the most devastating aspects of ageing, with no effective treatments available. Here we have defined a pathway linking intestinal P. goldsteinii, MCFAs, myeloid cell GPR84 signalling, cytokines TNF and IL-1β, vagal dysfunction and reduced hippocampal novelty responses, which contributes to declining memory function during ageing.
Our results provide several conceptual advances. Brain ageing has a large brain-extrinsic component. Although several examples of humoral factors involved in the regulation of brain ageing have been defined45,46, less is known about the neuronal body–brain signals that contribute to the decline in brain function. Our study suggests that interoceptive routes of communication between the gut and the brain may lose their function over the course of the lifespan, with reduced sensory input into the brain contributing to aspects of brain ageing. Thus, interoceptive dysfunction might constitute a common principle underlying several diseases of the ageing brain.
Our findings also indicate that interoceptive dysfunction is not only a passive process that develops over the lifespan, but is also actively influenced by alterations in the peripheral inflammatory milieu. Low-grade inflammation is a major driver of organismal ageing47 but it shows marked heterogeneity in the human population48. It is possible that P. goldsteinii and MCFAs contribute to this heterogeneity as one example of numerous inflammatory triggers that can instigate cognitive decline through the pathway described in this study. This environment-sensitive vagal-hippocampal circuit may thus serve as a common downstream element linking several previous observations, such as the role of peripheral macrophages in age-associated cognitive decline49, the impact of diet-induced microbiome changes on memory50, the cognitive sequelae of post-viral syndromes51 and the neuroprotective effects of incretin hormones52,53.
Finally, although interactions between myeloid cells and neurons in the brain are critical in the context of cognitive decline, our data suggest that inflammatory processes do not necessarily need to take place in the central nervous system to affect cognitive function. Thus, a local inflammatory milieu in a peripheral tissue, such as the gastrointestinal tract, might be sufficient to trigger sensory neuron dysfunction and cognitive sequelae even in the absence of a generally heightened inflammatory response detected in the blood or brain.
Several major challenges remain in understanding the details of the proposed pathway. First, the polysynaptic pathway linking the intestine to the hippocampus remains poorly defined. A previous report26 and our observations (Extended Data Fig. 4c,d) imply a possible role for the medial septum in brainstem–hippocampal communication. Second, the mechanisms by which chronic peripheral inflammation influences vagal activity need to be understood. The vagus nerve responds to IL-1β (ref. 54), but how chronic exposure to this cytokine may alter vagal excitability is unclear. Third, the type and location of peripheral myeloid cells that contribute to interoceptive dysfunction is unclear. Finally, it remains to be determined whether elements of the pathway described here in mice contribute to age-associated cognitive decline in humans. Vagus nerve stimulation has been reported to enhance recognition memory, reward motivation and cognitive performance during sleep deprivation55,56,57,58, but it needs to be evaluated whether vagus-directed strategies are suitable to counteract age-associated cognitive decline.
These open questions notwithstanding, our findings emphasize the importance of body-derived signals in cognition and highlight the ability of the interoceptive system to counteract age-associated decline in hippocampal function and short-term memory. Pharmacological activators of interoceptive pathways—which we refer to as interoceptomimetics59—may thus have the potential to stimulate sensory input into the brain to boost the formation of memory engrams in the hippocampus. Our findings call for the systematic exploration of possible interoceptomimetics and their impact on the ageing brain.
Unless noted otherwise, young female (8 weeks old) and male (4 weeks old) C57BL/6 mice were obtained from The Jackson Laboratory and old mice (18 months old) were acquired from the National Institute on Aging. For co-housing, mice were housed either two young + three old, or three young + two old per cage. Males were co-housed before puberty to avoid fighting. Non-co-housed control young and old mice had their cages mixed so that two mice were swapped between each cage to control for the social effect of cage mixing. Where possible, mice were randomized to experimental groups, and experimenters were blinded to the experimental condition. Animals were housed in facilities at the University of Pennsylvania, the Arc Institute or Stanford University in 7 am to 7 pm light–dark cycles, 20–25 °C and 30–70% humidity. All experimental procedures were performed according to approval by the IACUC committees at the University of Pennsylvania, Stanford University and the Arc Institute.
Germ-free C57BL/6 mice were maintained in sterile isolators at the University of Pennsylvania Gnotobiotic Animal Facility. For FMT, one stool pellet per recipient mouse was homogenized in 1.5 ml of sterile phosphate-buffered saline (PBS) in an anaerobic hood and filtered through 70 μm filters. This homogenate (200 μl) was orally gavaged into germ-free mice housed in positive pressure isocages; in addition to oral gavage, 50 ml of used bedding from donor cages was added to the isocages.
The following mouse strains were used in manuscript and were purchased from The Jackson Laboratory: C57BL/6J (000664), CD45.1 (002014), DBA/2J (000671), Trpv1Cre (017769), Snap25-GCaMP6s (025111), DTA (009669), Phox2bCre (016223), Phox2bFlpO (022407), Il1rfl/fl (028398), Ccr2−/− (004999), Nlrp3−/− (021302), Tnfr−/− (003243), Cre-dependent hM4Di (026219), Cre/FlpO-dependent hM4Di (029040) and Ai65 (Cre/FlpO-dependent tdTomato) (021875). Gpr84−/− mice were a kind gift from I. Kimura60.
Antibiotic treatment, bacterial colonization and stool collection
Mice were given a combination of neomycin (1 g l–1; Research Products International); ampicillin (1 g l–1; Research Products International); vancomycin (0.5 g l–1; Mylan); metronidazole (0.5 g l–1; Research Products International); imipenem or cilastatin (0.5 g l–1; Fresenius Kabi); and ciprofloxacin (0.2 g l–1; Sigma-Aldrich) in their drinking water for two weeks. Parabacteroides goldsteinii (ATCC) and Alistipes shahii (ATCC) were grown under anaerobic conditions at 37 °C in Tryptic Soy Broth (BD Biosciences) supplemented with 5% defibrinated sheep’s blood (Hardy Diagnostics). Lachnospiraceae bacterium (DSMZ) was grown under anaerobic conditions at 37 °C in fastidious anaerobe broth (FAB) (Neogen). Lactobacillus acidophilus (ATCC) and Escherichia coli K12 (ATCC) were grown under aerobic conditions according to supplier instructions. Bacteria were gavaged into mice after two weeks of antibiotics treatment 24 h after the cessation of treatment. Mice were gavaged every other day the first week, and then once a week after. Control mice received PBS gavage. Stool samples for all experiments were collected fresh in 1.7 ml Eppendorf tubes and immediately snap frozen on dry ice before storage at −80 °C until DNA extraction.
P. goldsteinii and A. shahii were grown for two days in 50 ml tubes before centrifugation for 20 min at 3,220 × g at 4 °C. The supernatant was sterilized through 0.45 μm filters before 30 min of centrifugation through a 3 kDa size filter (Thermo Fisher Scientific) at 3,220 × g at 4 °C. The <3 kDa fraction was then treated with 25 μg ml–1 proteinase K (Thermo Fisher Scientific) for 1 h at 37 °C and then boiled for 10 min at 99 °C. This solution (300 μl) was gavaged into mice for five days before testing 1 h after the final gavage.
Mice received 50 mg kg–1 bromodeoxyuridine (BrdU) (MedChem Express) dissolved in sterile PBS via intraperitoneal injection for five consecutive days, brains were collected four weeks after final injection.
Capsaicin, cholecystokinin, GLP1 agonist and CCKAR antagonist treatment
Capsaicin (Sigma-Aldrich) was dissolved at 25 mg ml–1 in 10% Tween-80, 10% ethanol and 80% PBS. Mice were intraperitoneally injected with 5 μg kg–1 capsaicin for five days before testing. Cholecystokinin octapeptide (2 μg kg–1 CCK; Bachem), GLP1 (amino acid positions 7–36) (4 mg kg–1, Cayman Chemical, 15069) and liraglutide (1 mg kg–1; Toronto Research Chemicals) were dissolved in PBS and injected 1 h before testing. The CCKAR receptor antagonists devazepide (MedChem Express) and A-65186 (Biorbyt) were dissolved in 10%:10% DMSO:Tween-80 in PBS and injected intraperitoneally for five days at 1 mg kg–1 before testing.
3-HOA (100 mg kg–1; 1PlusChem), embelin (100 mg kg–1; Ambeed), decanoic acid (20 mg kg–1; Sigma-Aldrich) and PBI-4050 (20 mg kg–1; TargetMol) were dissolved in 5% DMSO, 5% Tween-80 and 90% PBS. An oral gavage was conducted for five days, with 1 h after the final treatment. For water supplementation, decanoic and dodecanoic acid (Sigma-Aldrich) were dissolved at 100 mg ml–1 in Tween-80 and then added to drinking water at 0.2 mg ml–1 for ten days; daily water consumption was assumed to be 3 ml per mouse.
All antibodies were obtained from BioXCell. For cytokine depletion, mice received intraperitoneal injections of anti-TNF (150 μg, BP0058) or anti-IL-1β (50 μg, BE0246) on days 0, 4 and 7 before testing on day 8. For T cell depletion, mice received 200 μg intraperitoneal injections of anti-CD4 (BE0003) on days 0 and 4 and 200 μg anti-CD8α intraperitoneally (BE0061) on day 0 before testing on day 7. For natural killer cell depletion, mice received 200 μg intraperitoneal injections of anti-NK1.1 (BE0036) on days 0 and 4 before testing on day 7. For neutrophil depletion, mice received daily intraperitoneal injections of anti-Ly6G (25 μg, BP0075-1) and anti-rat immunoglobulin G (50 μg, BE0122) every other day for one week before testing61.


