
E. coli is a pervasive Gram-negative bacterium that colonizes nearly all individuals and is an important cause of serious invasive infection11,12,13. These considerations highlight unresolved questions regarding how colonization affects infection susceptibility. For extra-intestinal infections such as urinary tract or bloodstream infections, colonization by strains that express defined virulence factors such as fimbrial adhesins or capsular polysaccharides is often identified as an infection risk factor14,15,16. However, despite near ubiquitous colonization by strains capable of invasive infection, E. coli bacteraemia still occurs infrequently, ranging from 10–100 per 100,000 person-years in the general population17,18, suggesting that host-associated factors have more important roles, especially for individuals at high risk for invasive infection.
A provocative alternative consideration relates to increasingly recognized protective benefits associated with colonization by pathobiont microorganisms. Staphylococcus aureus bacteraemia-related death and all-cause mortality are significantly reduced among S. aureus-colonized individuals despite higher rates of nosocomial bacteraemia19. Natural antibodies—defined as those that are not related to obvious previous infection or immunization—against E. coli and other commensal bacteria have also been consistently identified in healthy individuals20,21 and mice22,23, suggesting that systemic immunogenicity primed by commensal colonization is the norm rather than the exception. Immunogenicity to E. coli can also be primed by colonization with other cross-reactive Gram-negative bacterial species in the larger Enterobacteriaceae family24,25. Natural antibodies induced by cross-reactive commensals protect against systemic E. coli and Salmonella infection23, and protect neonatal mice against intestinal enterotoxigenic E. coli after vertical transfer24. Reciprocally, deficiency of maternal antibodies to Group B Streptococcus is correlated with serotype-specific susceptibility of neonates to invasive infection26.
Although these findings suggest that defects in vertically transferred anti-E. coli natural immunity contribute to infection susceptibility in neonates, broader applicability remains uncertain as neither immunogenicity primed by E. coli colonization, nor antibody levels in human babies with E. coli infection, nor susceptibility to systemic infection by extra-intestinal pathogenic E. coli strains that cause neonatal sepsis have been evaluated. Here we leveraged emerging platforms that enable antibody recovery from dried blood spot specimens27, collected one day after birth for routine newborn screening, with preclinical models of E. coli preconceptual colonization and neonatal infection to demonstrate that early-life susceptibility reflects diminished quantity and functional opsonization activity of vertically transferred natural maternal antibodies.
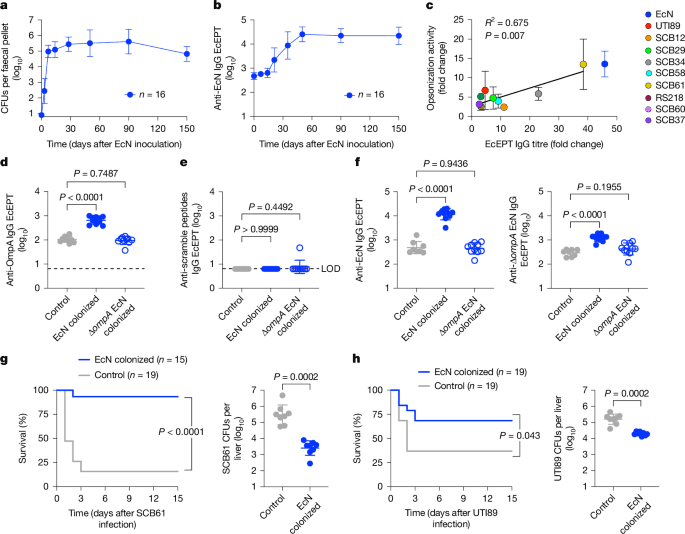
Mice reared under standard specific pathogen-free laboratory conditions exhibit facility-dependent variation and are often devoid of commensal E. coli, which limits translational applicability, considering that humans are ubiquitously colonized11,12,24. This shortcoming was bypassed by first establishing E. coli colonization, followed by investigating colonization-induced immunogenicity and susceptibility to extra-intestinal infection. We found that sustained intestinal colonization with the non-pathogenic human probiotic E. coli Nissle 1917 (EcN) strain is efficiently established and reaches a plateau in the faeces of adult mice without antibiotic treatment, within 7 days after a single oral inoculation10 (Fig. 1a). EcN colonization was limited to proximal and distal digestive organs, and spared normally sterile tissues including the spleen, liver and brain (Extended Data Fig. 1a). Nonetheless, colonization primed progressively increased serum anti-EcN IgG titres that plateaued by day 50 and were most enriched with IgG2b and IgG1 compared with other antibody isotypes, which were increased more modestly or remained at background levels similar to those of control mice without EcN colonization (Fig. 1b; Extended Data Fig. 1b,c).
a, EcN in the faeces after a single oral inoculation (2 × 106 CFU) to 6-to-8-week-old C57BL/6 wild-type mice under specific pathogen-free laboratory conditions. b, IgG end point EcEPT titres against EcN in the sera of mice described in a. c, Fold change in IgG titres compared with opsonization activity against the indicated E. coli isolates in the sera of mice 50 days after EcN colonization compared with control mice without E. coli colonization (n = 4 individual mice (EcN, SCB61); n = 12 (UTI89); n = 3 (SCB12, SCB29, SCB34, SCB58, RS218, SCB60, SCB37)). d,e, IgG titres that recognize five pooled OmpA outer loop peptides (d) compared with scramble control peptides (e) in mice 50 days after oral inoculation with EcN (n = 10) or ΔompA EcN (n = 10) compared with control mice without E. coli colonization (n = 10). f, IgG titres against EcN (left) or ΔompA EcN (right) for mice 50 days after oral inoculation with EcN (n = 10) or ΔompA EcN (n = 10) compared with control mice without E. coli colonization (n = 8). g,h, Survival and bacterial burden 1 day after intravenous infection with SCB61 (g) or UTI89 (h) for mice 50 days after oral inoculation with EcN (n = 8 (g); n = 7 (h)) or control mice without E. coli colonization (n = 8 (g); n = 7 (h)). Data are presented as mean ± s.d. Differences between groups were analysed using simple linear regression (c), one-way ANOVA (d–f), unpaired Student’s t-test (g,h) or log-rank (Mantel–Cox) test (g,h). LOD, limit of detection.
To investigate cross-reactivity to other E. coli strains, and particularly to clinically relevant isolates, sera from EcN-colonized mice were titred against a panel of E. coli clinical isolates. Given the importance of sequence types 95, 69, 131 and 1193 for causing neonatal sepsis28,29,30, we focused on clinical isolates of these sequence types, which have distinct O-antigen, K-capsule and flagella serotypes that were non-overlapping with EcN29,31,32,33,34,35,36 (Extended Data Fig. 1d). EcN colonization primed broad cross-reactivity with significantly increased IgG titres against each isolate in the sera of EcN-colonized mice compared with in control mice without EcN colonization (Extended Data Fig. 1e). Sera from EcN-colonized mice also efficiently opsonized these clinical isolates, with opsonization activity directly proportional to the IgG titre (Fig. 1c), highlighting functional activity against virulent E. coli by natural antibodies primed by commensal EcN.
To further investigate E. coli cross-reactive antigens, these analyses were extended to a panel of isogenic strains with targeted mutations in genes encoding serotype-defining cell-surface structures, including waaL (encoding a ligase required for O-antigen attachment to lipid A core), kpsD (encoding a transporter required for K-capsule presentation), fliC (encoding a major flagella structural protein) or rfaH (encoding a transcription elongation factor required for O-antigen and K-capsule expression)37,38. Despite loss of these serotype-defining antigens individually or in combination, cross-reactivity was preserved with similar increased anti-E. coli IgG in the sera of EcN-colonized mice when titred against the mutant strains compared with their respective parental wild-type strain (Extended Data Fig. 2).
Other targets of host immunity include ubiquitous outer membrane proteins, such as OmpA, which possesses multiple transmembrane domains and four outer surface loop domains35,39 (Extended Data Fig. 3a). OmpA immunogenicity was probed using five peptides, encompassing the outer loop 1, 2 and 4 domains plus two distinct loop 3 peptides, given the wider heterogeneity in these residues between EcN and clinical isolates that cause invasive infection (Fig. 1d and Extended Data Fig. 3a,b). EcN colonization induced sharply increased anti-OmpA loop peptide-specific IgG, which was overturned in mice colonized with ΔompA EcN (Fig. 1d and Extended Data Fig. 3b,c), and without cross-reactivity to scramble control peptides (Fig. 1e). Of note, sera from ΔompA EcN-colonized mice exhibit reduced wild-type EcN-specific IgG and lack ΔompA EcN-specific IgG, indicating that OmpA is not only a target, but is also essential for EcN-primed serological immunogenicity (Fig. 1f).
However, serological immunogenicity primed by commensal EcN is not exclusively targeted against OmpA, as titres against ΔompA EcN were still consistently increased (Fig. 1f and Extended Data Fig. 4). In turn, nearly identical increased IgG titres were found against other ΔompA variants of established clinical isolates compared with their respective parental wild-type strain (Extended Data Fig. 4). These findings align with serological cross-reactivity primed by EcN colonization to other Gram-negative Enterobacteriaceae with distinct OmpA outer loop peptides including Klebsiella, Yersinia, Citrobacter, Enterobacter and Salmonella species, but not against unrelated microorganisms, including Gram-positive S. aureus and Listeria monocytogenes bacteria or Candida albicans fungi (Extended Data Fig. 5a,b). Together, these findings identify OmpA as one of several serological targets for natural antibodies primed by commensal E. coli and as a necessity for E. coli colonization-induced immunogenicity.
Protection primed by EcN colonization was evaluated by comparing susceptibility of mice infected intravenously with virulent E. coli. We focused particularly on SCB61, which represents a strain with pronounced EcN cross-reactivity, and UTI89, which has reduced cross-reactivity and is widely used in preclinical studies6,31 (Fig. 1c and Extended Data Fig. 1e). These experiments showed significantly improved survival after infection with either SCB61 or UTI89, and reciprocally reduced bacterial burden in target tissues of EcN-colonized mice compared with in those of control mice without EcN colonization (Fig. 1g,h and Extended Data Fig. 6a). Improved survival and reduced pathogen burden were more pronounced after SCB61 infection than after UTI89 infection, consistent with relative levels of EcN serological cross-reactivity (Fig. 1g,h and Extended Data Figs. 1e and 6a). Comparatively, protection against UTI89 infection was abolished in ΔompA EcN-colonized mice lacking cross-reactive IgG (Extended Data Fig. 6b,c), in agreement with the necessity for OmpA in priming immunogenicity. Significantly reduced pathogen burden in EcN-colonized mice after infection with Klebsiella pneumoniae, but not L. monocytogenes, S. aureus or C. albicans, (Extended Data Fig. 6d–g) further highlights protection by cross-reactive antibodies, rather than trained innate immune components40.
To more definitively establish the necessity for antibodies in colonization-induced protection, susceptibility to invasive infection was evaluated in mice transferred with serum from EcN-colonized wild-type or B cell-deficient μMT−/− mice, or from wild-type control donors without EcN colonization. These experiments showed that antibodies are essential for colonization-induced protection, as only sera from EcN-colonized wild-type mice were able to transfer protection (Fig. 2a and Extended Data Fig. 6h). The importance of antibodies is further shown by the lack of EcN colonization-induced protection against UTI89 infection in μMT−/− mice compared with in wild-type mice (Fig. 2b and Extended Data Fig. 6i). Lack of protection in μMT−/− mice cannot be explained by inefficient EcN colonization given the similar colonization tempo and EcN recovery in the faeces (Extended Data Fig. 6j). Together, these findings demonstrate that commensal E. coli colonization primes natural antibodies that protect against invasive systemic infection.
a, Bacterial burden 1 day after SCB61 (left) or UTI89 (right) infection for mice administered with sera (100 μl) from control mice without EcN colonization (n = 8), wild-type (WT) mice on day 50 after EcN colonization (n = 8) or μMT−/− mice (n = 7) on day 50 after EcN colonization. b, Bacterial burden 1 day after UTI89 infection in wild-type mice compared with μMT−/− mice on day 50 after EcN colonization (n = 12 (wild type); n = 13 (μMT−/−)) or control mice without E. coli colonization (n = 12 (wild type); n = 11 (μMT−/−)). c, Bacterial burden 1 day after UTI89 infection (200 CFU intraperitoneal inoculation) in 3-day-old pups born to control mice without E. coli colonization (n = 9) compared with pups born to EcN-colonized wild-type (n = 10) or EcN-colonized μMT−/− (n = 5) dams. d, Bacterial burden 1 day after UTI89 infection in 3-day-old pups administered the indicated dosage of purified IgG from EcN-colonized or control mice without E. coli colonization 1 h prior to infection (n = 3 to 5 mice per group; each dot represents data from an individual mouse). e, Bacterial burden 1 day after UTI89 infection (left) or compared with anti-EcN IgG titres (right) for pups born to EcN-colonized dams (n = 10), pups born to control mice without E. coli colonization (n = 9) or cross-fostered within the first 12 h after birth (n = 9 (prenatal); n = 8 (postnatal)). f–h, Human anti-E. coli IgG levels in paired third trimester maternal specimens compared with cord blood after term pregnancy, titred against EcN (f), pooled neonatal infection clinical isolates RS218, SCB12, SCB29, SCB61, SCB34, SCB58, SCB37 and SCB60 (g) or a pool containing five OmpA outer loop peptides compared with scramble control peptides (h) (n = 10; lines indicate paired maternal–cord blood specimens). Data are presented as mean ± s.d. Differences between groups were analysed using one-way ANOVA (a,c,e (left)), unpaired Student’s t-test (b), simple linear regression (e (right)) or paired Student’s t-test (f–h).
Given pronounced early-life susceptibility and rising incidence of E. coli as a cause of severe infection in newborn babies1,2, this platform was used to further investigate whether preconceptual maternal EcN colonization can override neonatal susceptibility to E. coli infection. Neonatal mice are markedly more susceptible to relatively low inocula of UTI89 (200 colony-forming units (CFU)) administered by intraperitoneal inoculation than adult mice (Extended Data Fig. 7a). Despite this inherent early-life susceptibility, mortality and high tissue pathogen burden were reduced in 3-day-old pups born to EcN-colonized dams compared with age-matched control pups born to dams without EcN colonization (Fig. 2c and Extended Data Fig. 7b). Protection in neonatal mice requires maternal antibodies, as mortality and pathogen burden rebound in μMT+/− pups born to EcN-colonized μMT−/− dams, and to levels resembling those in pups born to dams without EcN colonization (Fig. 2c and Extended Data Fig. 7b). Protection was similarly observed after oral E. coli infection in pups born to EcN-colonized dams, representing a more relevant route of infection leading to systemic dissemination41 (Extended Data Fig. 7c).
Anti-E. coli IgG and IgG subclasses were efficiently transferred with similar titres in EcN-colonized dams and their offspring, compared with selectively reduced IgM in pups (Extended Data Fig. 7d), in agreement with inefficient placental transfer of this isotype42. Colonization-induced IgG antibodies are also sufficient for protection, given the reduced pathogen burden in neonatal mice administered low-dosage IgG from EcN-colonized donors compared with mice without E. coli colonization (Fig. 2d and Extended Data Fig. 7e). Vertically transferred protection is also not explained by commensal E. coli in-host evolution43, given the near-identical genomes of EcN recovered from colonized virgin or pregnant mice compared with the parental strain (Extended Data Fig. 7f). Thus, preconceptual E. coli colonization primes natural maternal antibodies that, upon vertical transfer, efficiently protect neonates against invasive systemic infection.
To further investigate when protective anti-E. coli IgG transfer occurs, pups born to EcN-colonized dams or to non-colonized control dams were cross-fostered within the first 12 h after birth, with infection occurring 3 days thereafter. These experiments showed progressively reduced susceptibility in pups with only prenatal exposure compared with those with only postnatal exposure, which were both significantly less susceptible than control pups with neither prenatal nor postnatal exposure to EcN-colonized dams (Fig. 2e and Extended Data Fig. 7g). Although these findings show that protection is transferred both in utero and after birth, postnatal exposure through breastmilk has a more prominent role, given the only modestly increased bacterial burden and marginally reduced anti-EcN IgG titres compared with pups born to and nursed by EcN-colonized dams (Fig. 2e and Extended Data Fig. 7g).
Applicability to human mother–infant dyads was evaluated by comparing anti-E. coli titres in paired third trimester maternal and cord blood specimens after term pregnancy (Extended Data Fig. 8a). This analysis showed similar anti-EcN IgG titres, including individual IgG subclasses, in cord blood compared with maternal blood specimen, whereas anti-EcN IgM titres declined precipitously (Fig. 2f and Extended Data Fig. 8b). IgG with specificity to E. coli clinical isolates that cause neonatal sepsis and OmpA were also efficiently transferred, as shown by similar titres against a mixture of the aforementioned eight neonatal sepsis clinical isolates containing two of each sequence type (Fig. 2g and Extended Data Fig. 1d), or OmpA loop peptides individually or pooled together in paired maternal–cord blood specimens compared with significantly reduced background titres against scramble control peptides (Fig. 2h and Extended Data Fig. 8c). Notably, as cord blood enables the contribution of in utero transfer to be evaluated separately and dissociated from transfer via breastmilk, these findings also demonstrate more efficient prenatal transfer of natural maternal IgG in human babies than in mouse pups, which are more reliant on postnatal acquisition of protective antibodies through breastmilk44,45.
To investigate how vertically transferred natural antibodies confer protection, we evaluated the effects of adoptively transferred serum from EcN-colonized adult donor mice on infection susceptibility in neonatal recipients. These experiments showed sharply reduced bacterial burden in E. coli-infected pups transferred with sera from EcN-colonized donors compared with sera from control mice without EcN colonization, and an essential role for antibodies, as protection was eliminated in pups transferred with sera from EcN-colonized μMT−/− donors (Fig. 3a and Extended Data Fig. 8d). This versatile platform was expanded to evaluate the requirement for defined antibody-interacting molecules by comparing how pups with targeted defects in these molecules respond to sera from EcN-colonized donors. These experiments show that complement and IgG Fc receptors (FcγR), including FcγRI, FcγRII, FcγRIII and FcγRIV46, are simultaneously essential, as protection conferred by sera from EcN-colonized wild-type donors is eliminated in FcγRnull, C3−/− and C1q−/− pups (Fig. 3b and Extended Data Fig. 8e). By contrast, CD22, which binds to sialylated glycoproteins including IgG47, is non-essential given the similar protection in Cd22−/− pups and in wild-type pups (Fig. 3b and Extended Data Fig. 8e).
a, Bacterial burden 1 day after UTI89 infection (200 CFU, intraperitoneal inoculation) in 3-day-old pups administered sera from control mice without EcN colonization (n = 8) compared with sera from EcN-colonized wild-type (n = 5) or EcN-colonized μMT−/− (n = 8) donors. b, Bacterial burden 1 day after UTI89 infection in 3-day-old pup or eight-week-old adult FcγRnull, C3−/−, C1q−/− or Cd22−/− recipients administered sera from EcN-colonized wild-type donor mice (n = 6 to 8 mice per group; each dot represents data from one mouse) compared with sera from control mice without E. coli colonization (n = 6 to 8 mice per group; each dot represents data from one mouse). c, EcN opsonization by sera from control mice without EcN colonization (n = 7) compared with sera from wild-type (n = 7), μMT−/− (n = 7) or C3−/− (n = 7) EcN-colonized donors in RAW264.7 mouse macrophage cells. d, EcN opsonization by sera from control mice without E. coli colonization (n = 6) compared with sera from EcN-colonized donors (n = 6) with anti-CD16 and anti-CD32 Fc blockade (anti-CD16/32) or rat IgG2b isotype control antibody in RAW264.7 mouse macrophage cells. e, Opsonization of E. coli (pooled mix of neonatal clinical isolates RS218, SCB12, SCB29, SCB61, SCB34, SCB58, SCB37 and SCB60) by cord blood sera from term pregnancy after heat inactivation, supplementation with IgG-depleted human sera, or with anti-human Fc block, compared with goat polyclonal IgG isotype control antibody in THP1 activated human macrophage cells (left) or HL60 human differentiated neutrophil cells (right) (n = 8 unique cord blood specimens per group (THP1 cells); n = 6 unique cord blood specimens per group (HL60 cells)). Data are presented as mean ± s.d. Differences between groups were analysed using one-way ANOVA (a,c–e) or unpaired Student’s t-test (b).
A similar approach comparing susceptibility of adult recipient mice transferred with sera from EcN-colonized donors was used to investigate whether this necessity for complement and FcγR is unique to the neonatal developmental window. Notably, and in sharp contrast to 3-day-old recipients, protection by sera from EcN-colonized mice persisted in adult 8-week-old FcγRnull, C3−/− and C1q−/− recipient mice (Fig. 3b and Extended Data Fig. 8e). These findings unveil developmental distinctions in how natural immunity works, with IgG being selectively reliant on opsonization through FcγR and complement in neonates. Together with diminished early-life complement levels and neutrophil activity48,49, these results further highlight that neonates have minimal excess capacity in these immune components compared with adults, with more substantial functional redundancy among protective antibody-interacting molecules.
We next investigated opsonization in vitro, where the individual contribution of sera containing natural IgG antibodies, complement and FcγR could be evaluated separately. These experiments verified the shared necessity for complement and FcγR for E. coli opsonization, as activity was only stimulated by sera from EcN-colonized wild-type mice, compared with background levels in sera from EcN-colonized μMT−/− or EcN-colonized C3−/− mice, which were similar to those in wild-type control mice without EcN colonization (Fig. 3c). Lack of opsonization by sera from EcN-colonized μMT−/− and EcN-colonized C3−/− mice reflects only the absence of antibody and complement, respectively, as combining sera from these mice with individual missing components restored opsonization activity (Fig. 3c). Similarly, FcγR blockade with anti-CD16/CD32 IgG overturned opsonization activity by sera from EcN-colonized wild-type mice in a dose-dependent manner50 (Fig. 3d). Opsonization by vertically transferred maternal antibodies was further evaluated in macrophage and neutrophil cells from human cord blood serum specimens, which showed similar reliance on complement and FcγR, as activity abolished by heat inactivation was efficiently restored with IgG-depleted human sera containing intact complement and neutralized with anti-human FcγR blocking IgG (Fig. 3e). Together, these findings demonstrate that natural antibodies protect neonates selectively through opsonization and vertical transfer of maternal IgG with anti-E. coli opsonization activity.
Anti-E . coli IgG in human neonatal sepsis
To further investigate whether E. coli infection in human neonates reflects inadequate vertically transferred natural maternal antibodies, anti-E. coli IgG titre and opsonization activity were evaluated in babies with neonatal sepsis utilizing banked dried blood spot specimens collected the first day after birth (median 25 h) for routine newborn screening (Fig. 4a). Babies with E. coli sepsis were identified retrospectively based on International Classification of Disease (ICD) diagnostic codes and were compared with three control babies without infection who were individually matched for sex, gestational age (±1 week) and birth timing (same month and year) to normalize these variables potentially affecting vertically transferred immunity (Fig. 4a). Specimens from 100 babies with neonatal E. coli sepsis showed similar sex distribution, with a wide distribution in gestational age (median 35 weeks, range 23 to 42 weeks) and infection age (median 4 days, range 1 to 29 days) (Fig. 4b). Although not a criterion used for selection or exclusion, none of these babies had galactosaemia, a known risk factor for E. coli sepsis that is identified by newborn screening51. Given the limited sample quantity and lack of information on particular E. coli features that cause infection in this retrospective platform, together with the established importance of E. coli sequence types 95, 69, 131 and 1193 in human neonatal sepsis28,29,30, anti-E. coli activity was evaluated using a mixture of the aforementioned 8 neonatal sepsis clinical isolates containing 2 of each sequence type (Extended Data Fig. 1d).
a, Dried newborn blood spot specimens were obtained from babies with E. coli neonatal sepsis and three control babies without infection who were matched for sex, gestational age and infection timing. Left, photo courtesy of Chris Lima, March of Dimes. Middle, BioTrust logo reprinted with permission from MDHHS Communications. Right, ELISA plate schematic created in BioRender; Way, S. S. https://biorender.com/wz9mo0w (2026). b, Birth gestational age and infection onset age distribution among 100 babies with neonatal E. coli sepsis. c, Anti-E. coli IgG titres in newborn blood spots from babies with E. coli sepsis (n = 100) compared with the average in matched control babies (n = 100) titred with pooled neonatal infection clinical isolates (RS218, SCB12, SCB29, SCB61, SCB34, SCB58, SCB37 and SCB60) evaluated by birth gestational age. d, IgG levels in newborn blood spots from babies described in c (n = 100 (E. coli sepsis); n = 100 (averaged control babies)). e,f, IgG titres in newborn blood spots against five pooled OmpA loop peptides (e) or scramble control peptides (f) for babies described in c (n = 100 (E. coli sepsis); n = 100 (averaged control babies)). g,h, Anti-E. coli opsonization activity for newborn blood spot specimens against eight pooled neonatal infection clinical isolates in THP1 activated macrophage cells (g) or HL60 differentiated neutrophil cells (h) (n = 100 (E. coli sepsis); n = 100 (averaged control babies)). Each dot represents data from an individual blood spot specimen from babies with E. coli sepsis (red) or the average from three matched control babies for each case (blue); the shaded region indicates 95% confidence intervals. Data are mean ± s.d. Differences between groups analysed using unpaired Student’s t-test (b, gestational weeks), Mann–Whitney U test (b, infection onset age), paired Student’s t-test (c–h), or regression intercepts using analysis of covariance (c–h).
These experiments showed consistent tenfold reductions of anti-E. coli IgG in blood spots from babies with neonatal sepsis compared with the average titre in control babies without infection who were matched for sex, gestational age and birth timing (Fig. 4c). Reductions in anti-E. coli IgG primarily reflect diminished IgG2 titres, which showed even more exaggerated (more than 40-fold) reductions between specimens from babies with E. coli sepsis compared with specimens from control babies without infection, whereas anti-E. coli IgG1 did not differ significantly, and IgG3 and IgG4 titres were only inconsistently detected (Extended Data Fig. 9a). These reductions in anti-E. coli IgG are not explained by diminished overall antibody recovery, given the similar gestational age-dependent changes in total IgG and IgG subclasses between babies with E. coli sepsis and control babies without infection (Fig. 4d and Extended Data Fig. 9b,c).
Blood spots from babies with neonatal sepsis also contain reduced anti-OmpA IgG, albeit of lower magnitude than with detection using whole E. coli bacteria (Fig. 4c,e), in agreement with the aforementioned preclinical data highlighting OmpA as an important target for E. coli serological immunogenicity. Use of scrambled control peptides confirmed the specificity of neonatal IgG for binding to OmpA outer loop peptides (Fig. 4f). Given the importance of natural antibody opsonization for host defence in neonates, opsonization activity was further evaluated in these newborn blood spot specimens. As our focus was investigating opsonization differences due to anti-E. coli antibodies, IgG-depleted human sera containing complement was supplemented so that potential changes in opsonization activity could be directly attributed to differences in antibodies that recognize E. coli (Fig. 3e). These experiments showed consistently diminished anti-E. coli opsonization activity in macrophage and neutrophil cells for specimens from babies with E. coli sepsis compared with the average activity in specimens from matched control babies without infection across gestational ages (Fig. 4g,h).
Stratifying anti-E. coli titres and IgG-dependent opsonization activity across birth gestational age further clarifies that overlap in absolute titres and opsonization activity between babies with E. coli sepsis and control babies without infection primarily reflect reductions in overall IgG levels in babies born preterm, shown by the significant reductions in anti-E. coli and anti-OmpA IgG titres and opsonization activity across gestational ages (Fig. 4c–h). Risk reduction cut-offs for each assay were also identified using conditional logistic regression to evaluate the distribution of titres and opsonization activity without consideration for gestational age (Extended Data Fig. 10a–d). Particularly for this cohort, which contains 100 babies with E. coli neonatal sepsis and 296 matched control babies without infection, anti-EcN IgG end point titres (EcEPTs) below 2,500 or anti-OmpA EcEPTs below 1,000 would predict around 20% risk of neonatal sepsis (Extended Data Fig. 10a–d), which is significantly higher than current epidemiological estimates1,2 of around 1 case per 1,000 live births or 0.1% risk.
Postnatal age of infection onset within the neonatal window is another parameter that is consistently related to E. coli susceptibility1,2. Evaluating this parameter as an independent variable showed that reductions in anti-E. coli IgG, anti-OmpA IgG or IgG-dependent opsonization activity, along with similar overall IgG levels and titres against anti-OmpA scramble control peptides, are maintained in babies with E. coli sepsis regardless of age at infection onset (Extended Data Fig. 10e–j). For babies with early-onset sepsis, diminished anti-E. coli IgG titre and opsonization were also not explained by antibody adsorption to bacteria as shown by comparable magnitude reductions in anti-E. coli, anti-OmpA IgG titre and opsonization activity in babies with E. coli sepsis identified within the first two days after birth compared with infection identified after three days of age (Extended Data Fig. 10k). Together with the efficiency whereby vertically transferred maternal IgG overrides the susceptibility of neonatal mice to E. coli infection, these findings demonstrate diminished vertically transferred natural immunity in the form of anti-E. coli antibodies and their opsonization function as universal risk factors for neonatal sepsis.
Neonates are highly susceptible to E. coli, with severe infections occurring in up to 1 in every 1,000 live births1,2. However, infections might be expected to occur even more frequently given ubiquitous exposure with transfer of pathogenic maternal strains shortly after birth3,4,5,52,53, together with hypo-responsiveness and skewed tissue distribution of neonatal immune cells6,7,8,9. Our studies investigating why most babies do not develop E. coli sepsis further amplify the translational importance of previous studies investigating E. coli immunity using preclinical infection models by verifying the necessity of vertically transferred antibodies that recognize E. coli23,24. We find newborn blood spots from human babies with E. coli sepsis have significantly reduced anti-E. coli IgG titres and IgG-dependent anti-E. coli opsonization activity (Fig. 4c–h) that parallel the susceptibility of neonatal mice born to dams devoid of commensal E. coli colonization-induced IgG (Fig. 2c and Extended Data Fig. 7b). Restoring anti-E. coli IgG through vertical transfer from E. coli-colonized dams or horizontal transfer from E. coli-colonized donors efficiently protects neonatal mice against infection (Figs. 2c and 3a), resembling the tenfold increase in anti-E. coli IgG titre found in sex and gestation age-matched babies without infection (Fig. 4c).
Vertical IgG transfer increases throughout gestation, peaking in the third trimester, and early preterm birth is a significant risk factor for neonatal E. coli infection1,2,54. This progressive transfer of maternal IgG, previously detected primarily in cord blood, was recapitulated in our analysis of blood spots from babies on the basis of gestational age (Fig. 4d and Extended Data Fig. 9c). Although total IgG levels were generally lower among infants born before 32 weeks of gestation, those with E. coli sepsis still had reduced anti-E. coli IgG and opsonization activity compared to gestational age-matched control individuals regardless of gestational age at birth (Fig. 4c–h). Isolated deficiency in anti-E. coli IgG, rather than diminished total IgG levels, as the primary risk factor for neonatal sepsis further suggests that systemic invasion occurs much more frequently, with clinical infection developing primarily in babies without sufficient vertically transferred anti-E. coli IgG to eliminate invasive bacteria. In turn, levels of anti-E. coli IgG or IgG-dependent opsonization below defined thresholds shortly after birth can be used to identify babies at highest risk regardless of gestational age at birth (Extended Data Fig. 10a–d). Indeed, the lower anti-E. coli IgG levels in babies with E. coli sepsis suggests the possibility that defects may exist in preconceptual maternal colonization, colonization-primed serological immunity, vertical transfer of these natural protective maternal antibodies or a combination of these factors. Although individually these defects are likely to occur infrequently, their combined frequency is expected to be approximately 1 in 1,000, similar to the proportion of newborns who develop E. coli sepsis.
These results support a general adoption of prenatal screening for anti-E. coli IgG similar to current evaluations of expecting mothers for serological immunity against pathogens such as tetanus or varicella, to which babies are particularly vulnerable. Targeting E. coli OmpA, and especially outer domains of this transmembrane protein that is found in all E. coli, may be especially informative given the reduced titres to these defined antigens in blood spot specimens from babies with E. coli sepsis (Fig. 4e). Comparing anti-OmpA titres in nearly 400 blood spot specimens (100 from babies with E. coli sepsis and 296 from matched control babies without infection) identifies levels of anti-OmpA and anti-E. coli IgG along with IgG-dependent anti-E. coli opsonization above which risk of sepsis is significantly decreased (Extended Data Fig. 10a–d). Important next steps include comparing anti-OmpA and anti-E. coli IgG in mothers of babies with E. coli sepsis, along with levels of these antibodies in paired maternal–neonate samples, similar to studies on Group B Streptococcus26.
Our finding that antibodies that recognize E. coli are prevalent in healthy babies regardless of gestational age, together with preclinical data showing that anti-E. coli IgG efficiently protects neonatal mice, whether delivered parenterally or through breastmilk, also raises the exciting prospect of preventing infection in infants at high risk using immunoglobulin enriched with anti-E. coli antibodies administered systemically or via early-life feeding. Averting infection with immunoglobulin may extend to other pathogens responsible for neonatal sepsis, including K. pneumoniae, given the reduced IgG with this specificity associated with neonatal sepsis55. Finally, considering broad cross-reactivity primed by EcN colonization against E. coli clinical isolates that cause neonatal sepsis, exploring use of this probiotic strain in women of reproductive age or expecting mothers to reduce E. coli neonatal infections, similar to the way in which maternal immunization against influenza, COVID-19 or tetanus can reduce neonatal infection56,57,58,59, represents compelling actionable next steps. Unlike subunit vaccines that are currently being developed for E. coli, which prime immunity against a limited set of defined antigens35,60, immunogenicity primed by commensal EcN expressing a more diverse array of endogenous protective antigens with cross-reactivity to virulent strains regardless of O-antigen, K-capsule and flagella serotypes would be expected to confer broader protection. Alternatively, given the OmpA immunogenicity associated with protection against systemic infection in both preclinical infection models and natural infection of babies resulting in neonatal sepsis, further investigations on the use of OmpA and other E. coli immunodominant outer membrane proteins as candidate vaccine antigens are warranted.
Inherent limitations associated with retrospective identification of neonatal E. coli sepsis and associated use of de-identified dried blood spot specimens collected for newborn screening include the lack of maternal prenatal specimens and microbiological data on the exact E. coli strain that caused sepsis in each individual, and whether infected neonates had other immunological or anatomic risk factors. These considerations prevent evaluation of maternal E. coli colonization as well as strain-specific virulence and antigenic features of pathobiont E. coli, which should also be a focus of follow-up studies to further investigate the link with diminished vertically transferred IgG in neonatal E. coli sepsis. Additional next steps for realizing these translational considerations include prospectively evaluating these parameters in larger human maternal–infant birth cohorts that are sufficient to capture the approximate 1 case in around 1,000 live births. Nonetheless, reframing how neonatal E. coli infection is conceptualized, focusing on resistance instead of susceptibility for the majority of babies, and incorporating critical elements related to vertically transferred natural maternal immunity yields insights into unique mediators of early-life immunity with important implications for understanding protection against other human pathobionts.
Wild-type mice on the C57BL/6 background and transgenic strains including µMT−/− (B6.129S2-Ighmtm1Cgn/J; 002288), Cd22−/− (C57BL/6-Cd22tm1Lam/J; 006940), C3−/− (B6.129S4-C3tm1Crr/J; 029661) and C1q−/− (B6(Cg)-C1qatm1d(EUCOMM)Wtsi/TennJ; 031675) were purchased from The Jackson Laboratory61,62,63,64 and maintained under specific pathogen-free housing conditions at Cincinnati Children’s Hospital. FcγRnull (FcRα null) mice with combined defects in FcγRI, FcγRII, FcγRIII, FcγRIV were provided by J. Ravetch46. All mice were housed under specific pathogen-free conditions in individually ventilated cages with ad libitum food (tekland 2919) and ultrafiltered UV-sterilized water under controlled environmental conditions including 22 °C, humidity 30–80% and 14 h–10 h light–dark daily cycles. Sex and age-matched groups of adult (6–8 weeks) mice were randomized for EcN colonization or no colonization control groups, and monitored for up to 150 days thereafter. For cross-fostering, pregnant mice were checked for delivery at least every 12 h, and pups were transferred to dams with or without EcN colonization after birth. Infected mice were checked at least every 12 h and euthanized when moribund for survival outcome experiments. All animal studies were performed under Cincinnati Children’s Hospital Research Foundation IACUC approved protocols.
Features for E. coli strains Nissle 1917 (EcN), UTI89, RS218, SCB12, SCB29, SCB61, SCB34, SCB58, SCB37 and SCB60 are described in Extended Data Fig. 1d. EcN and other parental wild-type E. coli strains including EC958, MS7163 and RS218 (refs. 65,66,67; Extended Data Fig. 4a) and mutants with targeted defects in serotype-defining cell-surface structures (waaL, kpsD, fliC, rfaH or ompA) were generated by λ-Red-mediated homologous recombination as described37,38,68. Reference wild-type K. pneumoniae (43816), Yersinia enterocolitica (JB580v), Citrobacter koseri (BAA-895), Enterobacter cloacae (13047), Salmonella typhimurium (ST14028), L. monocytogenes (10403 s), S. aureus (USA200) and C. albicans (SC5314) have been described69,70,71,72,73,74,75. For oral inoculation, EcN was cultured overnight in brain–heart infusion (BHI) liquid medium, sub-cultured to early log-phase growth (OD600 0.1; 37 °C; 200 rpm), washed and diluted in sterile saline and administered dropwise into the mouth of unanaesthetized adult mice (2 × 106 CFU in 50 µl). For infection, bacterial strains were cultured overnight in BHI liquid medium, sub-cultured to early log-phase growth (OD600 0.1; 37 °C; 200 rpm), washed and diluted in sterile saline and administered intravenously via the lateral tail vein. Adult mice were infected with E. coli (5 × 106 CFU for bacterial burden quantification; 4 × 107 CFU for survival outcomes). Three-day-old neonatal mice were infected with E. coli (200 CFU in 50 µl, intraperitoneally or 5 × 106 CFU, oral gavage) as described6,76. Other bacteria including K. pneumoniae, L. monocytogenes and S. aureus were administered at the following dosages intravenously (1 × 105 CFU K. pneumoniae; 1 × 105 CFU L. monocytogenes; 7.5 × 106 CFU S. aureus in 200 µl) for infection in adult mice. For fungal infection, C. albicans was cultured overnight in yeast extract peptone adenine hemisulfate dextrose (YPAD) liquid medium, sub-cultured early log-phase growth (OD600 0.1; 30 °C; 200 rpm), washed and diluted in sterile saline and administered intravenously to adult mice (1 × 106 CFU) as described77. The bacterial and fungal inoculum in each experiment was verified by spreading serial dilutions onto media agar plates. For enumerating the number of recoverable E. coli, K. pneumoniae, L. monocytogenes, S. aureus or C. albicans from colonized or infected mice, the faeces or tissue was dissected in sterile conditions and homogenized in saline containing 0.05% Triton X-100. Serial dilutions of the faeces or tissue homogenates were spread onto media agar plates, and the number of individual colonies enumerated after incubation at 37 °C for 24 h.
Blood collected from adult EcN-colonized or control mice via retroorbital or cheek bleeding, or from neonatal mice after decapitation, was allowed to clot at room temperature for 20 min and then centrifuged for serum collection. For adoptive transfer, sera from donor mice were pooled and transferred intraperitoneally to 8-week-old adult (100 µl) or 3-day-old neonatal (50 µl) recipient mice 4 h prior to infection. For IgG purification, frozen serum was thawed and purified using NAb Protein A Plus Spin Column (Thermo Fisher Scientific), quantified on the basis of optical density (NanoDrop), verified for purity by protein gel electrophoresis and Coomassie Blue staining, resuspended in sterile saline and administered intraperitoneally to 3-day-old neonatal recipient mice 1 h prior to infection.
Paired maternal and cord blood specimens after term pregnancy were collected through the IMPRINT cohort designed to investigate the effect of initial respiratory virus exposures on infant immunity78. This cohort began in October 2019 after receiving Institutional Review Board approvals from Cincinnati Children’s Hospital Medical Center and the University of Cincinnati Medical Center (IRB 2019-0629 and SITE00000489, respectively). Women were enroled in the third trimester of pregnancy, with collection of maternal sera and cord blood collected at delivery. Additional non-paired cord blood specimens from healthy donors were provided in a de-identified manner through Cincinnati Children’s Hospital Applied Gene and Cell Therapy Center, Cell Processing Facility. For newborn blood spots, 100 babies with neonatal sepsis were identified through International Classification of Diseases diagnostic codes ICD9-03842 and ICD10-P36.4 corresponding to ‘E. coli septicemia’ and ‘sepsis of the newborn due to E. coli’, respectively. ICD9-03842-based identification was limited to individuals ≤28 days old to identify neonatal infection (Fig. 4b). Three control babies without infection were individually matched for each case with identical sex, gestational age (±1 week) and birth timing (same month and year), except for two babies with E. coli sepsis born at 23 weeks of gestation, where only one matched control baby could be identified, were identified through the Michigan BioTrust for Health79. Three 6-mm circular punches from dried blood specimens for each baby with E. coli sepsis and corresponding control babies without infection were provided in a de-identified manner. All studies were performed under Cincinnati Children's Hospital Medical Center and Michigan Department of Health and Human Services IRB approved protocols.
Antibodies against E. coli and other bacteria and fungi were titred using high-binding 96-well immunoassay plates (Corning, 9018) coated with microorganisms from log-phase (OD600 0.5–0.7) growth, washed and diluted in saline and allowed to dry overnight under UV light. Thereafter, bacteria or fungi-coated plates were blocked with milk (3%), serum titred starting at a 1:10 dilution with 5 additional 1:5 serial dilutions and probed with biotin-conjugated secondary antibodies including: rat anti-mouse IgG (ThermoFisher 13-4013-85 (RRID AB_466650); 1:2,000 dilution), rat anti-mouse IgM (ThermoFisher 13-5890-82 (RRID AB_466761); 1:2,000 dilution), rat anti-mouse IgA (ThermoFisher 13-5994-82 (RRID AB_466863); 1:2,000 dilution), rat anti-mouse IgE (BD Pharmingen 553419 (RRID AB_394850); 1:2,000 dilution), rat anti-mouse IgG1 (BD Pharmingen 553441 (RRID AB_394861); 1:2,000 dilution), rat anti-mouse IgG2b (BD Pharmingen 553393 (RRID AB_394831); 1:2,000 dilution), rabbit anti-mouse IgG2c (ThermoFisher SA5-10235 (RRID AB_2810193); 1:2,000 dilution) and rat anti-mouse IgG3 (BD Pharmingen 553401; RRID AB_394838; 1:2,000 dilution).
For human anti-E. coli antibodies, maternal or cord blood serum specimens were titred starting at a 1:10 dilution with 5 additional 1:5 serial dilutions and probed with biotin-conjugated secondary antibodies including: goat anti-human IgM (ThermoFisher A18841 (RRID AB_2535618); 1:2,000 dilution), mouse anti-human IgG1 (BD Pharmingen 555869 (RRID AB_396187); 1:2,000 dilution), rabbit anti-human IgG2 (ThermoFisher MA5-27931 (RRID AB_2744977); 1:2,000 dilution), mouse anti-human IgG3 (ThermoFisher MH1532 (RRID AB_2539711); 1:2,000 dilution) and mouse anti-human IgG4 (BD Pharmingen 555882 (RRID AB_396194); 1:2,000 dilution). Overall human IgG levels were evaluated by pooling antibodies against each IgG subclass at 1:2,000 dilution. For dried blood spots, each 6-mm punch, estimated to contain 5 μl of serum, was dissolved overnight at room temperature in 100 µl of sterile saline with gentle shaking80,81,82 and titred starting at a 1:10 dilution with 5 additional 1:5 serial dilutions. Total human antibody levels were measured using the following commercial kits according to the manufacturer’s recommendations: human IgG (Thermo Fisher Scientific, 88-50550), human IgM (Thermo Fisher Scientific, 88-50620-88), human IgG1 (Thermo Fisher Scientific, 88-50560-22), human IgG2 (Thermo Fisher Scientific, 88-50570-22), human IgG3 (Thermo Fisher Scientific, 88-50580-22) and human IgG4 (Thermo Fisher Scientific, 88-50590-22). Plates were developed with streptavidin–peroxidase (BD Bioscience 554066; 1:1,000 dilution) using o-phenylenediamine dihydrochloride as a substrate and measuring absorbance at 450 nm. Titres were quantified as end point titre (EcEPT), the reciprocal of the highest analyte dilution that gives a reading above the cut-off, which was set to 0.1 using a non-linear second order polynomial regression as described47.
For evaluating antibodies against OmpA, each peptide (loop 1, WSQYHDTGFINNNGPTHEN; loop 2, GRMPYKGSVENGAYKA; loop 3a, KSNVYGKN; loop 3b, KANVPGGASFKD; loop 4, TNNIGDAHTIGTRPDNGM; loop 1 scramble, TQPNSHNDINTNHGYGWE; loop 2 scramble, RSMAGKGPKNAGYVYE; loop 3a scramble, KYNGVNSK; loop 3b scramble, FKAKDNGVGSPA; loop 4 scramble, MTNTHNTAIGDRNIGPGD) at >90% purity (GenScript) was diluted in saline for coating 96-well (600 ng per well; for titrating mouse sera) or 384-well (120 ng per well; for titrating human sera or eluted dry blood spots) ELISA plates overnight at 4 °C. For probing the response to multiple peptides, equal amounts of each peptide, 600 ng per well (96-well pates) or 120 ng per well (384-well plates), were utilized. Thereafter, peptide-coated plates were blocked with 2% PBSA, and serum or eluted dry blood spots were titred starting at a 1:10 dilution with 5 additional 1:5 serial dilutions and probed with the aforementioned biotin-conjugated anti-mouse IgG and anti-human IgG secondary antibodies.
To enumerate cell-associated bacteria, E. coli strains were transformed with GFP-expressing plasmid pBbB5c-GFP (Addgene, 35353)83 and maintained in medium supplemented with chloramphenicol (20 µg ml−1). For opsonization by mouse sera, RAW 264.7 mouse macrophage (ATCC TIB-71) cells (2 × 105 cells per well) were seeded in flat-bottom 96-well cell culture plates (Cellstar, 655180) and allowed to adhere for 1 h achieving >80% confluency. Thereafter, the medium was exchanged with 30 µl of saline containing mouse sera at a 1:5 dilution plus GFP+ E. coli (106 CFU per well) and allowed to incubate at 37 °C for 30 min. After incubation, the plate was washed three times with sterile saline to eliminate non-adherent bacteria and macrophage-adherent bacteria were measured by fluorescence (OD530 after 480 nm excitation). For some experiments, the GFP+ E. coli serum mixture was supplemented with rat anti-mouse CD16/32 (clone 2.4G2; BioXcell BE0307 (RRID AB_2736987)) or rat IgG2b kappa isotype control (clone LTF.2; BioXcell BE0090 (RRID AB_1107780)). For opsonization by human sera or blood spot specimens, THP1 activated macrophage (ATCC TIB-202) cells (1 × 104 cells per well), cells were seeded into 384-well tissue culture treated plates (Corning, 3701) to >80% confluency in DMEM medium supplemented with recombinant human TGFβ1 (1 ng ml−1; R&D, 7754-BH) and 1α,25-dihydroxyvitamin D3 (50 nM; Millipore Sigma, 705942) for 24 h. Adherent cells were then stimulated with phorbol-myristate acetate (15 ng ml−1; Sigma-Aldrich) for 15 min for macrophage differentiation as described84. For neutrophil cells, human myeloid leukaemia HL60 (ATCC CCL-240) cells (1 × 104 cells per well) were seeded into 384-well tissue culture treated plates (Corning, 3701) in RPMI 1640 medium supplemented with l-glutamine, penicillin–streptomycin, HEPES, 10% FBS and 1.3% DMSO for 5 days as described85. Thereafter the medium was exchanged with 8 µl of saline containing human serum or blood spot elution at a final 1:40 dilution plus pooled GFP+ E. coli (2 × 105 total CFU per well), which were allowed to incubate at 37 °C for 30 min. After incubation, the plate was washed three times with sterile saline to eliminate non-adherent bacteria, and adherent bacteria were measured by fluorescence (OD530 after 480 nm excitation). RAW 264.7, THP1 and HL60 were routinely tested (about every six months) for mycoplasma negativity. Besides confirmation based on cellular morphology and species-specific phagocytic function after incubation with mouse or human antibodies, no additional cell line authentication was performed. To inactivate complement, serum was heat-inactivated (56 °C, 30 min) prior to use. For some experiments, the serum:bacteria mix was supplemented with 20% immunoglobin depleted human serum as a source of complement (Celprogen, HS2001).


