
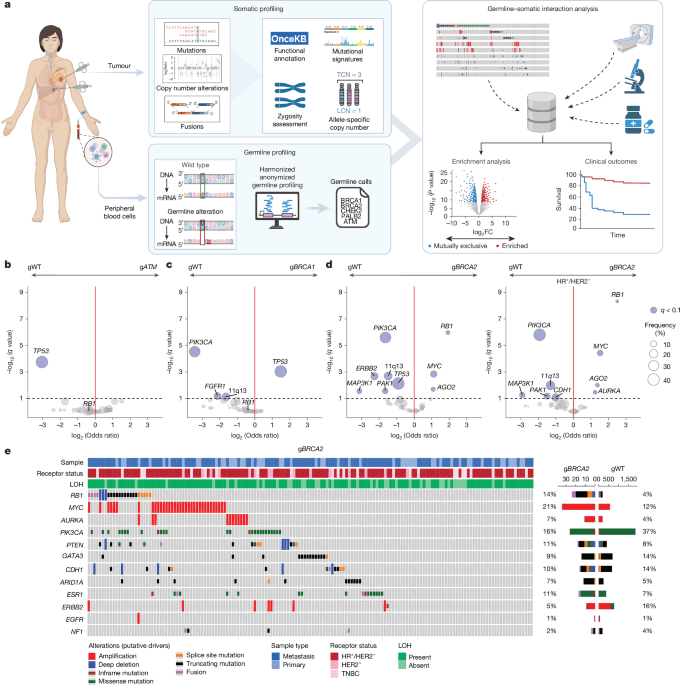
Resistance to anticancer therapy often occurs through a diverse array of genomic mechanisms1,2,3,4,5. The inherent unpredictability of these events challenges the ability to develop proactive strategies to pre-empt the emergence of resistance and improve patient outcomes. The role of germline pathogenic variants in predisposition to malignancies and shaping the initial somatic landscape of tumours is well established6,7,8,9. This interplay is well exemplified in germline pathogenic variants in certain DNA damage repair genes10,11,12,13,14 that give rise to characteristic somatic allelic configurations and genomic instability in the form of homologous recombination deficiency (HRD)15,16,17, a biology that has been successfully exploited with PARP inhibitors (PARPi)18,19,20. However, despite our understanding of these initial events, the influence of germline pathogenic variants on the subsequent evolutionary life of a tumour remains poorly defined.
Clinically, the expanding landscape of approved targeted and lineage-directed therapies has introduced considerable uncertainty regarding the optimal treatment paradigm for patients with breast cancer with certain germline and somatic backgrounds. Given its dominant effect on initial cancer evolution, we posit that the germline background may equally influence tumour behaviour under therapeutic pressure, effectively directing the evolutionary trajectory of disease progression and resistance.
More broadly, defining how pre-treatment germline and somatic genomic context shapes the path of acquired therapy resistance could enable early interception strategies to prevent or delay the emergence of drug-resistant tumour clones. To elucidate the therapeutic relevance of germline–somatic interactions in a clinically meaningful setting, we performed an integrated analysis of germline and somatic genomic data paired with detailed clinical annotation, including treatment response, in a large cohort of patients with breast cancer.
Clinical and genomic features of the cohort
To identify the interactions between germline pathogenic variants and somatic oncogenic alterations in breast cancer, we integrated detailed clinical annotation with prospectively collected sequencing data21,22,23 based on 6,927 tumours from 5,881 patients with breast cancer (Memorial Sloan Kettering Cancer Center (MSK) cohort; Extended Data Fig. 1a and Supplementary Table 1). DNA derived from tumour tissue and blood as a source of germline DNA were each sequenced using an FDA-authorized clinical sequencing assay encompassing up to 506 cancer-associated genes, including germline analysis of 84 cancer predisposition genes21,22,23. Genes of interest for the germline analysis included canonical members of the homologous recombination pathway10,11,12,13,14: BRCA2 (2.9%, n = 161 patients), BRCA1 (2.6%, n = 142 patients), CHEK2 (1.6%, n = 87 patients), ATM (1.1%, n = 60 patients) and PALB2 (0.6%, n = 35 patients).
The clinicopathological characteristics of the germline-altered cancers and germline wild-type (gWT) cancers strongly reflected previously established patterns, suggesting that our cohort was representative of the broader population of patients with breast cancer (Table 1). Specifically, we observed a younger age of diagnosis in gBRCA1/2 carriers than in gWT. gBRCA1-associated breast cancers tended to be triple-negative24 and high-grade invasive ductal carcinomas. Meanwhile, gBRCA2, gCHEK2 and gATM carriers typically had hormone receptor-positive and HER2-negative (HR+/HER2–) disease (75.2%, 74.7% and 71.7%, respectively; Supplementary Table 2), consistent with previous studies25,26.
Biallelic loss is often a necessary condition to observe a phenotypic impact in carriers of germline pathogenic variants in HRD-related genes, yet its incidence varies by gene, with higher rates of biallelic inactivation observed in high-penetrance genes27,28. Our results confirmed that biallelic inactivation rates varied significantly across genes, ranging from 50.6% (n = 44) in gCHEK2 carriers to 77.5% (n = 110) and 75.8% (n = 122) for gBRCA1 and gBRCA2 carriers, respectively (Extended Data Fig. 1b). We also found lower frequency of loss of heterozygosity (LOH) in gPALB2 carriers (51.4%, n = 18), but a relatively higher frequency of ‘second-hit’ somatic mutations resulting in biallelic loss (33.3%, n = 6), concurring with previous literature29. Confirming the associations between histological, demographic and genomic patterns with germline pathogenic variants establishes the clinical relevance of this clinicogenomic cohort.
We first sought to define patterns of mutual exclusivity or enrichment of somatic variants with germline pathogenic variants (germline–somatic interactions), in the context of breast cancer receptor subtype and zygosity (Fig. 1a). This analysis robustly validated previously reported enrichment of TP53 alterations in gBRCA1 carriers and mutual exclusivity of gATM and TP53 alterations30,31 (Fig. 1b,c). Both findings were more pronounced in tumours exhibiting biallelic inactivation of the respective genes (Extended Data Fig. 2a,e). We further explored these observations by conducting germline–somatic interaction analyses stratified by breast cancer receptor subtypes. The gBRCA1–TP53 interaction was enriched in HR+/HER2– tumours but not in triple-negative tumours, where TP53 variants are already highly prevalent in BRCA1 WT tumours, supporting TP53 loss of function (LoF) as a ubiquitous event in the oncogenesis of gBRCA1-driven breast cancers32,33.
a, Study schema. Enrichment analysis was performed to identify somatic alterations more prevalent in patients with pathogenic germline variants of homologous recombination pathway genes (compared with ‘sporadic’ or gWT cases). FC, fold change; LCN, lesser copy number; TCN, total copy number. Schematic created in BioRender; Razavi, P. https://biorender.com/hv0bvbr (2025). b, Enrichment analysis of gATM (n = 60) compared with gWT (n = 5,094), as determined by Firth-penalized logistic regression with sample type and receptor status as covariates. Significant genes (blue) are defined by q < 0.10. c, As in panel b, but for comparison of gBRCA1 (n = 142) with gWT (n = 5,904).gBRCA1 tumours were enriched for somatic TP53 variants; OR = 2.80 (95% CI 1.70–4.59, q = 0.0009) and mutually exclusive with PIK3CA variants. d, As in panels b,c, but for comparison of gBRCA2 (n = 161) compared with gWT (n = 5,094). A subset analysis of HR+/HER2– tumours (nTotal = 3,547, ngBRCA2 = 121 and ngWT = 3,426) was then performed. Somatic RB1 variants represented the most significant enrichment in gBRCA2 carriers among this subgroup; OR = 5.66 (95% CI 3.34–9.60, q = 4.7 × 10−9). e, Oncoprint showing mutations, copy number deletions and fusions in the indicated genes in gBRCA2 versus gWT. Receptor status, sample type and zygosity are annotated above.
In gBRCA2-driven breast cancers, RB1 somatic variants were significantly enriched, in stark contrast to their absence in gBRCA1-associated tumours (Fig. 1c–e and Extended Data Fig. 2b). This observation is notable, as gBRCA1 tumours are largely triple negative, a subtype typically enriched for RB1 alterations. Focusing on patients with HR+/HER2− tumours (75.2% of gBRCA2) revealed an even higher enrichment of somatic RB1 alterations in gBRCA2 carriers. This analysis also uncovered an enrichment of MYC and AURKA amplifications. Of clinical relevance, RB1, MYC and AURKA alterations have all been implicated in resistance to CDK4/6 inhibition34,35. Conversely, PIK3CA alterations were more enriched in gWT cancers than in gBRCA2 and gBRCA1-driven cancers. The receptor status and LOH-specific germline–somatic interactions are further displayed in Extended Data Fig. 2, and corresponding Oncoprints are detailed in Extended Data Fig. 3 (Supplementary Tables 3–7).
CDK4/6i combined with endocrine therapy (CDK4/6i + ET) represents the cornerstone of treatment for patients with metastatic and high-risk early-stage HR+/breast cancer36,37,38, with RB1 loss established as a key mechanism of resistance to CDK4/6i39. On the basis of our results demonstrating a significant enrichment of RB1 alterations in gBRCA2 carriers, we analysed the effect of gBRCA2 status on progression-free survival (PFS) in patients with HR+/HER2– metastatic breast cancer (MBC) treated with CDK4/6i + ET. gBRCA2 pathogenic variants were associated with a significantly shorter PFS on CDK4/6i + ET in univariate and multivariate analyses (median PFS of 9.0 versus 15.6 months, multivariate hazard ratio (HR) = 2.17, 95% CI 1.60–2.96, P < 0.00001; Fig. 2a). Similar results were seen when the analysis was extended to all treatment lines, with consideration of ET partner and treatment line as covariates (HR = 1.97, 95% CI 1.55–2.51, P < 0.00001; Fig. 2b and Supplementary Table 8). LoF mutations in RB1 were rare in pre-treatment samples (2%), and exclusion of these cases did not alter the results.
a, PFS for patients treated with first-line CDK4/6i + ET by gBRCA2 status. Patients with gBRCA2 (n = 51) were compared with gWT (n = 926); P = 8.0 × 10−7. b, As in panel a, but depicting all lines of treatment. Patients with gBRCA2 (n = 80) were compared with gWT (n = 1,670); P = 2.9 × 10−8. c, PFS on first-line CDK4/6i + ET combinations by gBRCA2 status from the multi-institutional external validation cohort, which included 61 patients with the gBRCA2 pathogenic variant and 2,097 with gWT. HRs in panels a–c were estimated using Cox proportional hazard models (P values from two-sided Wald tests). No multiple comparisons adjustment was made. d, Swimmer’s plot of patients (n = 41) receiving PARPi (blue bar) after CDK4/6i + ET (red bar); HRPARPi versus HRCDK4/6i = 0.38 (95% CI 0.19–0.76, P = 0.0062), based on Cox proportional hazard model stratified by patient ID. Best response (complete or partial response) was compared for PARPi versus CDK4/6i + ET using a two-sided Fisher’s exact test; OR = 8.87 (95% CI 2.84–27.98, P = 0.00002). MD, physician; NE, non-evaluable. e,f, Two representative cases demonstrating short PFS on first-line (1L) CDK4/6i + ET and subsequent metabolic complete response (CR) and durable disease control on PARPi. 2L, second line; 3L, third line; AI, aromatase inhibitor; POD, progression of disease; PR, partial response. Black and yellow arrows denote radiographic tumour activity. g, Paired pre-CDK4/6i or post-CDK4/6i oncoprint by gBRCA2 status. The black rectangle denotes acquired variant. Two-sided Fisher’s exact test of RB1 LoF variant acquisition by gBRCA2 status; OR = 5.17 (95% CI 2.07–13.0, P = 0.0010).
We next validated our findings using an independent, nationwide clinicogenomic dataset containing manually curated patient-level outcomes data from both community oncology settings and academic medical centres40,41. This analysis confirmed a strong association between gBRCA2 and shorter PFS on CDK4/6i + ET (median PFS of 12.0 versus 18.3 months, HR = 1.83, 95% CI 1.36–2.48, P < 0.00001; Fig. 2c) among the 2,158 patients with HR+/HER2– MBC treated with first-line CDK4/6i + ET combinations. To evaluate whether gBRCA2 was associated with resistance specifically to CDK4/6i + ET, we expanded our analysis to assess the effect of gBRCA2 status on PFS of other common therapeutic modalities in breast cancer (Extended Data Fig. 4a). gBRCA2 status did not affect outcome on the vast majority of these therapies, nor did it significantly effect overall survival for patients starting on first-line CDK4/6i + ET. This demonstrates that gBRCA2 status is not a universal determinant of outcome but instead confers context-dependent relevance to specific therapies.
We further investigated the MSK cohort focusing on patients with HR+/HER2– MBC who received a PARPi after progression on CDK4/6i (n = 41). Of note, despite being administered in later lines (median line of therapy = 3), HRD-directed therapy generally resulted in superior outcomes (Fig. 2d). PARPi treatment resulted in a significantly improved PFS compared with the preceding CDK4/6i regimen (HR = 0.38, 95% CI 0.19–0.76, P = 0.0062), with 73.2% of patients achieving a longer PFS on PARPi than on frontline CDK4/6i. This clinical benefit was more pronounced among the patients who had failed to respond to previous CDK4/6i + ET (Fisher’s exact test; OR = 8.87, 95% CI 2.84–27.98, P = 0.00002; Extended Data Fig. 4b). Among patients with evaluable imaging who did not discontinue therapy due to early toxicity (n = 38), PARPi achieved a partial or complete response in 84.2% (n = 32) of patients, compared with only 39.5% (n = 15) for previous CDK4/6i + ET (Extended Data Fig. 4c). Representative cases of rapid progression through first-line CDK4/6i + ET, followed by prolonged complete response to PARPi are highlighted in Fig. 2e,f. Collectively, these results provide a clinical rationale for prioritization of PARPi for this genomically defined subgroup of patients with breast cancer with expected poor outcomes on CDK4/6i-based combinations.
To investigate the implications of germline status on the development of RB1 loss, an established mechanism of CDK4/6i resistance, we assembled a large cohort of patients with paired tumour samples collected pre-CDK4/6i and post-CDK4/6i (1,312 tumour and 513 plasma circulating tumour DNA (ctDNA) samples from 546 patients). Genomic analysis of this paired pre-treatment and post-progression cohort demonstrated that acquired somatic RB1 LoF alterations were significantly more prevalent in gBRCA2 tumours versus gBRCA2 WT tumours (28.6% versus 7.1%, OR = 5.17, 95% CI 2.07–13.0, P = 0.0010; Fig. 2g). The clinical responses and evolutionary trajectories of patients treated with CDK4/6i are highlighted by two representative patients with gBRCA2 HR+/HER2– MBC (Extended Data Fig. 4d,e). In both cases, acquired polyclonal RB1 LoF variants emerged post-CDK4/6i, underscoring a strong and specific evolutionary pressure for RB1 LoF mutations as a dominant mechanism of CDK4/6i resistance in these gBRCA2 tumours.
Baseline allelic state forecasts RB1 loss
Consideration of the genomic architecture of BRCA2-driven tumours provides further insight into the acquisition of RB1 LoF alterations. BRCA2 and RB1 are syntenic on chromosome 13q, and previous work suggests that biallelic inactivation of BRCA2 in gBRCA2-driven tumours often occurs through loss of a large chromosomal segment inclusive of WT BRCA2 and RB1 alleles42,43,44,45,46,47. Consistent with this, the pattern of RB1 LOH was influenced by gBRCA2 status in our cohort, demonstrating a co-occurrence of BRCA2 and RB1 LOH in gBRCA2 tumours compared with gWT (Fig. 3a). We validated this finding using an external cohort of 46 gBRCA2-associated primary breast cancers profiled with whole-exome sequencing (24 patients from the University of Pennsylvania Abramson Cancer Center and 22 from the Mayo Clinic). Consistently, 82.6% (n = 38) of tumours in this cohort demonstrated concurrent LOH of BRCA2 and RB1 (P < 0.0001; Extended Data Fig. 5). Although RB1 LOH was enriched in gBRCA2 tumours, it notably was also present in 34.9% of gWT tumours (n = 601 of 1,723 of evaluable cases), suggesting a broader relevance for this allelic state.
a, RB1 and BRCA2 LOH in gBRCA2 (n = 63; top) versus gWT (n = 527; bottom); OR = 11.5 (95% CI 6.16–22.2, P < 2.2 × 10−16). b,c, PFS on first-line CDK4/6i + ET (b) and OS (c) by RB1 LOH, with numbers of LOH and no LOH given below. d,e, PALOMA-3 PFS (d) and OS (e) by RB1 LOH; OS P = 1.5 × 10−6. The numbers of LOH and no LOH are given below. f, Allelic configurations. g, Paired pre-CDK4/6i or post-CDK4/6i oncoprint (nRB1-Het loss = 145, nOther-LOH = 63 and nHeterozygous = 197); ORHet loss versus heterozygous = 11.5 (95% CI 3.50–46.1, P = 1.9 × 10−6) and OROther-LOH versus heterozygous = 3.22 (95% CI 0.57–18.21, P = 0.16). h, RB1 het loss predisposes to RB1 ‘second hit’. i, OS and PFS on first-line CDK4/6i + ET by RB1 ASCN (nRB1-Het loss = 180, nRB1-Other-LOH = 83 and nHeterozygous = 284). Error bars denote 95% CI. FGA, fraction genome altered; WGD, whole-genome duplication. j, Select RB1 variants in gBRCA2 tumours, short indels flanked by microhomology. Schematics created in BioRender: f, Razavi, P. https://biorender.com/utmr5e5 (2025); h, Razavi, P. https://biorender.com/bewn6wg (2025); j, Razavi, P. https://biorender.com/gxjrb6s (2025). k, HRD-associated RB1 variants (short indels and structural variants (SVs)) by gBRCA2; OR = 5.36 (95% CI 1.76–16.58). CN, copy number. l, PFS on first-line CDK4/6i + ET by RB1 ASCN and gBRCA2; HRgBRCA2 P = 1.2 × 10−10; numbers are given below. m, Acquired RB1 versus other TSG LoF. HRD association with non-RB1 TSG LoF: OR = 2.99 (95% CI 1.03–9.34, P = 0.045). ORs by two-sided Fisher’s exact test; HRs by multivariate Cox proportional hazard models (P from two-sided Wald test). No multiple comparisons adjustment were made. NS, not significant.
On the basis of our BRCA2–RB1 co-LOH results, we hypothesized that RB1 LOH is a predominant predisposing factor for CDK4/6i resistance. Hence, we extended our survival analyses to assess whether pre-treatment RB1 zygosity was predictive of clinical benefit from CDK4/6i beyond the context of gBRCA2 status. Specifically, we analysed 547 patients with HR+/HER2– MBC in the MSK cohort who received first-line CDK4/6i and had evaluable LOH status in the pre-treatment tumours. RB1 LOH was observed in 47.8% of these patients and was predictive of both significantly shorter PFS (HR = 1.53, 95% CI 1.24–1.89, P = 0.00007) and overall survival (OS; HR = 1.31, 95% CI 1.04–1.64, P = 0.022; Fig. 3b,c and Supplementary Table 9).
To confirm these findings, we performed an analysis of pre-treatment ctDNA samples collected as part of the PALOMA-3 study48, the pivotal randomized phase III trial of palbociclib plus fulvestrant versus fulvestrant monotherapy. RB1 LOH was observed in 19 (7.3%) of 259 pre-treatment ctDNA samples and was associated with a significantly shorter PFS (HR = 2.20, 95% CI 1.26–3.82, P = 0.0055) and OS (HR = 3.28, 95% CI 2.02–5.32, P < 0.00001; Fig. 3d,e). These trends were more pronounced in the palbociclib plus fulvestrant arm (Extended Data Fig. 6).
We further reasoned that the specific number of functional pre-treatment RB1 alleles influences the likelihood of acquiring a second-hit RB1 LoF alteration under the therapeutic pressure of CDK4/6i therapy. Specifically, we hypothesized that tumours with a single functional RB1 copy — reflecting RB1 hemizygosity (heterozygous loss (het loss)) — would be statistically more likely to develop an RB1 LoF variant as a second hit to achieve biallelic inactivation and resistance. To test this, we investigated the clinical relevance of pre-treatment RB1 het loss, as compared with other allelic configurations such as loss before or after whole-genome doubling resulting in more than one remaining WT RB1 alleles (Fig. 3f). We extended our analysis of pre-CDK4/6i and post-CDK4/6i tumours (n = 405 patients) to these refined RB1 allele-specific copy number (ASCN) definitions. Consistent with our hypothesis, only pre-treatment RB1 het loss uniquely predisposed to acquisition of RB1 LoF upon exposure to CDK4/6i (P < 0.00001; Fig. 3g). Acquired RB1 LoF events occurred almost exclusively in tumours with baseline RB1 het loss (total, 15.2%; acquired, 14.5%), whereas they were rare in other allelic settings. This suggests that RB1 hemizygosity lowers the evolutionary barrier to resistance, requiring only a single additional hit to achieve complete inactivation, whereas other allelic configurations require multiple independent events, making resistance via this route far less likely (Fig. 3h). To underscore RB1 loss as a specific mechanism of resistance to CDK4/6i, we leveraged our MSK cohort to categorize each sequenced sample by previous therapeutic exposure. Among all treatment classes (including ET monotherapy, chemotherapy, antibody–drug conjugates and targeted therapies), only CDK4/6i exposure was significantly associated with the enrichment of acquired RB1 LoF variants (Extended Data Fig. 7).
To distinguish between predictive versus prognostic properties of these changes, we repeated the outcome analyses based on the redefined pre-treatment RB1 allelic state (Extended Data Fig. 8). Both het loss (32.7%, n = 181) and other LOH allelic configurations (15.0%, n = 83) were associated with worse PFS on CDK4/6i. As chromosomal instability is a known poor prognostic factor49, we repeated this analysis adjusting for fraction genome altered and whole-genome doubling. Of note, RB1 het loss was the sole significant allelic configuration associated with shorter PFS in this multivariate analysis. Conversely, other LOH configurations were uniquely associated with worse OS (HR = 1.96, 95% CI 1.45–2.66, P = 0.00001; Fig. 3i and Supplementary Table 8), suggesting they represent a more general prognostic marker, whereas RB1 hemizygosity specifically serves as a predictive marker of outcomes on CDK4/6i.
Together, although the acquisition of RB1 LoF variants conferring resistance to CDK4/6i is relatively uncommon in an unselected population50 (8.9% in our paired pre-CDK4/6i-treated and post-CDK4/6i-treated cohort), our results demonstrate that acquired RB1 loss is both prevalent and highly predictable in tumours with a single functional RB1 allele before treatment. These findings have major clinical implications for the development of novel surveillance strategies and for patient selection for clinical trials of precision therapeutic strategies directed against complete RB1 loss51,52. More broadly, our results suggest that the pre-treatment allelic structure can forecast not only therapeutic outcomes but also the specific molecular trajectories through which targeted therapies fail.
We next assessed whether gBRCA2-mediated HRD specifically contributes to somatic RB1 LoF variants during CDK4/6i therapy. Hence, we analysed the types of alterations in RB1 found in our gBRCA2 cohort. We observed that somatic RB1 variants identified in gBRCA2 tumours predominantly exhibited the characteristic pattern of microhomology flanking a short deletion, a hallmark of back-up DNA repair mechanism through microhomology-mediated end joining in HRD-driven tumours53 (Fig. 3j). Indeed, we observed significant enrichment for short indels or structural variants within the coding sequence of RB1 in the gBRCA2 setting, indicative of BRCA2 deficiency-mediated lack of competent DNA repair (Fig. 3k).
Integration of our genomic analysis with treatment response data also suggested that HRD-mediated mutagenesis was probably responsible for the exceptionally rapid progression observed in gBRCA2 carriers treated with CDK4/6i combinations. Although RB1 LOH was associated with a significantly shorter PFS in gWT cases (HR = 1.47, 95% CI 1.19–1.82, P = 0.0004), in the context of gBRCA2, RB1 LOH resulted in an even shorter PFS (HR = 3.05, 95% CI 2.17–4.29, P < 0.00001; Fig. 3l). Thus, gBRCA2 breast cancers with RB1 LOH appear to exploit the imperfections of back-up DNA repair mechanisms to escape therapeutic pressure. This phenomenon resembles the concept of ‘exaptation’, which refers to an evolutionary co-option of a trait to serve a purpose distinct from its original role54.
We expanded our analysis to assess the role of HRD (of either germline or somatic origin) and clinical outcomes on CDK4/6i. The presence of a HRD-associated signature in pre-treatment tumours of patients with HR+/HER2– MBC conferred a significantly worse outcome on CDK4/6i + ET (HR = 1.78, 95% CI 1.27–2.48, P = 0.00075), even outside the context of gBRCA2 (Extended Data Fig. 9). Having established that this phenomenon extends beyond BRCA2, we next investigated whether a similar HRD-driven second-hit pattern was evident across other tumour suppressor genes (TSGs) implicated in CDK4/6i resistance. Non-BRCA2 HRD tumours showed a significantly increased tendency to acquire resistance via TSGs other than RB1 (37.5% of cases, OR = 2.99, 95% CI 1.03–9.34, P = 0.045) relative to non-HRD cases. The majority of cases with these acquired TSG LoF alterations were preceded by het loss of the respective gene in the pre-treatment setting (Fig. 3m).
In summary, the data suggest that both an increased prevalence in RB1 LOH and ongoing HRD-driven mutagenesis, which hastens the acquisition of RB1 LoF mutations, contribute to the poor outcomes of patients with breast cancer with gBRCA2 mutations treated with CDK4/6i + ET. Our findings demonstrate that this concept is not limited to gBRCA2 and RB1, but rather reflects a broader principle linking HRD biology and baseline allelic configurations to vulnerabilities across TSGs implicated in therapy resistance.
To directly validate the role of Rb loss as a driver of CDK4/6i resistance in gBRCA2-associated tumours, we generated several patient-derived xenografts (PDXs) from BRCA2 carriers with HR+/HER2– breast cancer. The first set of PDXs was derived from a 30-year-old gBRCA2 pathogenic variant carrier with de novo MBC treated at MSK (Fig. 4a). Clinically, this patient received abemaciclib in combination with pembrolizumab in the second-line setting as part of a trial, followed by standard-of-care CDK4/6i + ET with rapid disease progression. A biopsy of the rapidly progressing chest wall lesion revealed an RB1 LoF variant, which was absent in the pre-CDK4/6i metastatic biopsy. This patient subsequently received radiation therapy to the progressive chest wall disease concurrently with systemic platinum, resulting in a major response. This patient then received treatment with PARPi, until resistance emerged through acquisition of a BRCA2 reversion variant (c.3640_3855del). Despite several additional lines of cytotoxic chemotherapy, the patient eventually succumbed to the disease and participated in the MSK Last Wish Program (MSK IRB Protocol #15-021), enabling a rapid autopsy and post-mortem tissue collection and analysis.
a, PDXs derived from two metastatic deposits in a rapid autopsy study of an MSK patient with gBRCA2: PDX-R and PDX-L. ADC, antibody–drug conjugate; ICI, immune checkpoint inhibitor. Schematic created in BioRender; Razavi, P. https://biorender.com/gjwhomb (2025). b,c, In vivo efficacy study of PDX-L (n = 5; b) and PDX-R (n = 4; c) treated with vehicle, fulvestrant (200 mg kg−1 subcutaneous, twice weekly), ribociclib (75 mg kg−1 PO 5 days per week), or fulvestrant and ribociclib. PO, oral dosing. P = 1.29 × 10–13, as calculated by a linear mixed-effect model. Data are mean ± s.e.m. d, Western blot analysis of PDX-L and PDX-R with vehicle and ribociclib (n = 3 replicates). Actin was used as a loading control. e, Schematic representing the RB1 structural variant from whole-genome sequencing of a CDK4/6i-resistant PDX-L sample, resulting in deletion of exons 5–17. Chr. chromosome. f, PDX-C was derived from a patient with gBRCA2. Vehicle was compared with palbociclib (75 mg kg−1 PO 5 days per week). Data of individual mice are shown to demonstrate that tumours developing relative resistance to palbociclib also harboured Rb loss, as demonstrated in the western blot in panel g (n = 3 replicates). h, In vivo efficacy study of PDX-P1. PDXs were treated with vehicle, camizestrant (10 mg kg−1 PO daily), palbociclib (50 mg kg−1 PO daily), saruparib (1 mg kg−1 PO daily) or combination (camizestrant + palbociclib and camizestrant + saruparib). PO dosing was discontinued following takedown of the vehicle arm. Experiments included n = 5 mice in the palbociclib ± camizestrant arms and n = 10 mice in all other arms. P = 1.55 × 10–8. i, In vivo efficacy study of PDX-P2, with arms as defined in panel h. Experiments included n = 7 mice in the camizestrant monotherapy arm and n = 8 mice in all other arms. P = 2.72 × 10–77. For panels h,i, data are mean ± s.e.m.; P values were determined by two-way repeated measures analysis of variance (ANOVA) followed by Sidak’s multiple-comparisons test. Dotted horizontal line indicates tumour volume = 0.
Multi-site sequencing of autopsy samples did not reveal any evidence of previously detected RB1 somatic variant by deep targeted next-generation sequencing panel testing, indicating that this specific subclone was probably eradicated by subsequent therapies received after CDK4/6i exposure (Extended Data Fig. 10a and Supplementary Table 10). However, PDXs generated from two separate metastatic deposits in the lungs provided a unique opportunity to interrogate the evolutionary consequences of active HRD in the context of gBRCA2 and RB1 LOH. One PDX demonstrated BRCA2 reversion variant (PDX-R), whereas the other had persistent biallelic BRCA2 loss and no evidence of reversion variant on deep sequencing (PDX-L; Extended Data Fig. 10b). Otherwise, PDX-L and PDX-R displayed virtually identical copy number patterns, reflecting a strong genomic concordance between the two samples (Extended Data Fig. 10c).
When challenged with therapy, PDX-R tumours responded to ribociclib-based therapy, whereas PDX-L tumours were uniformly resistant (Fig. 4b,c). In the resistant PDX-L tumours, we observed no BRCA2 or Rb expression, whereas expression of both proteins was retained in sensitive PDX-R tumours (Fig. 4d). Whole-genome sequencing of CDK4/6i-resistant tumours from PDX-L identified a large structural variant in RB1, characterized by deletion of exons 5–17 (Fig. 4e). This acquired structural variant, combined with pre-existing RB1 het loss, resulted in complete loss of RB1. Such large deletions are a recognized hallmark of HRD-mediated instability55, highlighting the role of HRD in facilitating RB1 loss and validating our clinicogenomic observations in a rigorously controlled preclinical setting.
We next validated these findings in another PDX derived from a gBRCA2 carrier (PDX-C). The majority of samples developed resistance to palbociclib monotherapy (Fig. 4f), and Rb loss of expression was again observed in post-treatment tumours that were resistant to CDK4/6i, but not in CDK4/6i-sensitive tumours (Fig. 4g). The convergence of our clinicogenomic observations and the evidence from preclinical models implicating HRD in CDK4/6i resistance led us to compare the differential therapeutic response patterns between CDK4/6i and PARPi. In two additional independent gBRCA2 PDXs (PDX-P1 and PDX-P2), we demonstrated clear resistance in each CDK4/6i ± ET condition (palbociclib or camizestrant monotherapy and combination therapy), whereas sensitivity to selective PARP1 inhibition was maintained (saruparib monotherapy as well as in combination with camizestrant and fulvestrant; Fig. 4h,i).
Overall, these preclinical models validate our clinicogenomic findings, demonstrating that acquired RB1 loss underlies resistance to CDK4/6i in gBRCA2 tumours with active HRD. In this context, biallelic RB1 inactivation and Rb loss emerge under CDK4/6i selective pressure, whereas sensitivity to PARPi is maintained.
We have described disease-specific and subtype-specific patterns of germline–somatic interactions and their clinical implications in a large clinicogenomic cohort of patients with breast cancer. Patients harbouring gBRCA2 mutations or HRD arising from other causes experience a significantly worse outcome on combination CDK4/6i + ET, the current frontline standard of care in the metastatic HR+/HER2– setting. This finding was validated using a large, independent, multi-institutional real-world dataset representative of the general population of patients with MBC treated in both community and academic settings.
We have demonstrated a proclivity for gBRCA2 tumours to develop RB1 biallelic loss as a mechanism of resistance to CDK4/6i-based combination therapy and propose two separate characteristics of gBRCA2 tumours that facilitate the acquisition of RB1 loss. First, gBRCA2-driven breast cancers are more likely to harbour RB1 het loss before the start of CDK4/6i; we showed that this allelic configuration independently predisposes the tumour to loss of the unaltered RB1 allele. Second, the mutagenic processes active in HRD-driven tumours promote the acquisition of RB1 LoF variants as a mechanism of biallelic inactivation (a second hit). Consistent with these clinicogenomic findings, our preclinical models provide direct evidence mechanistically linking HRD and gBRCA2 to the acquisition of RB1 inactivation under the therapeutic pressure of CDK4/6i. In multiple gBRCA2 PDXs, acquired RB1 loss emerged as the dominant mode of CDK4/6i resistance in the context of active HRD. Conversely, sensitivity to PARPi was preserved, underscoring persistent vulnerability despite the emergence of CDK4/6i resistance.
These observations raise provocative questions regarding the optimal therapy sequence for gBRCA2-driven breast cancers. PARPi represent a proven targeted strategy for patients with HRD tumours20,56. However, given the long-standing use of CDK4/6i combinations as frontline standard of care for HR+ breast cancers, use of PARPi is typically relegated to later lines of therapy following the emergence of resistance to CDK4/6i + ET. Our data reveal a potentially greater benefit for PARPi therapy before CDK4/6i + ET in this molecularly defined subset of patients with breast cancer. The dominant mechanism of resistance to CDK4/6i + ET may reduce the utility of several therapies typically used later in the disease course that rely on an intact G1 checkpoint. As the development of RB1 loss appears to be partially mediated by HRD, earlier use of a PARPi may suppress this specific route of drug resistance resulting in a longer duration of response to subsequent CDK4/6i combinations. Specifically, reversion mutations that restore homologous recombination proficiency represent a widely prevalent and established mechanism of resistance to PARPi in gBRCA2 carriers57. Therefore, tumour cells with somatic reversion mutations in BRCA2 may be less likely to develop HRD-driven, RB1 LoF mutations under the selective pressure of subsequent CDK4/6i.
The insights gleaned from our analysis should prompt a re-evaluation of conventional clinical practice in which the sequence of therapies is dictated by the clinical benefit observed in a biomarker unselected population. Most relevant to our current study, our results question whether frontline PARPi therapy in patients with pathogenic gBRCA2 variants or HRD HR+/HER2– tumours would not only improve frontline outcomes but also establish the genomic prerequisites for more successful subsequent lines of therapy. Our data and this hypothesis serve as the impetus for the EvoPAR-Breast01 study (NCT06380751), a randomized international phase III clinical trial, which will compare frontline saruparib (a PARP1-selective inhibitor) plus camizestrant (an oral selective ER degrader) versus standard-of-care CDK4/6i + ET in patients with HR+/HER2– MBC harbouring germline or somatic alterations in selected HRD genes58.
More generally, we have demonstrated that pre-treatment RB1 het loss (hemizygosity) predicts the development of a somatic LoF variant involving the WT allele under the selective pressure of CDK4/6i. This proposed mechanism is the obverse of Knudson’s two-hit model, which seminally demonstrated that copy number loss occurs as a necessary second hit for oncogenesis in patients with hereditary retinoblastoma9. By contrast, in our proposed model, pre-existing RB1 het loss serves as the first hit increasing the likelihood of developing biallelic RB1 loss via acquired RB1 LoF variants as the second hit. Further studies will be needed to better refine the genomic and transcriptomic contexts predictive of CDK4/6i resistance resulting from biallelic RB1 inactivation.
Our study has potential limitations. First, we focused our analysis on exonic somatic variants that have previously demonstrated functional significance, which may fail to capture potentially consequential interactions, such as intronic variants59 or epigenetic silencing60 of RB1. Second, given the real-world nature of this retrospective analysis, the timing between a particular event (start of treatment or disease progression) and a particular biopsy varied. In addition, our paired pre-CDK4/6i and post-CDK4/6i analysis may have failed to capture clonal expansion events that were extinguished by further therapy. Despite these caveats, we were able to conclusively demonstrate that pre-treatment RB1 allelic configuration and HRD predict for the development of a second hit resulting in complete RB1 loss and our preclinical models provide strong evidence for biological plausibility of the proposed mechanism.
From a translational perspective, this work contributes to ongoing efforts to hasten the identification and interception of acquired resistance mechanisms. The ability to predict the precise mechanism of acquired resistance from genomic characteristics detectable in the pre-treatment sample would be a valuable tool in the precision oncology arsenal. Although our work principally illustrates this concept through RB1 hemizygosity, we hypothesize that the same framework may extend to other acquired tumour suppressors implicated in resistance. Currently defined ‘targetable’ genomic lesions encompass mutations, copy number homozygous deletions and amplifications, and structural variants, which in their present genomic configuration have already disrupted a specific oncogenic pathway. With this work, we expand the paradigm of the ‘actionable genome’ to states of allelic imbalance that have no biologic activity in their current allelic configuration but are predictive of therapeutic failure, and thus could be used to guide early interception strategies designed to delay the onset of resistance.


