
Biological research spans scales from molecules to systems to organisms, seeking to understand and design functional components across all domains of life. Creating a machine to design functions across the diversity of life would require it to learn a deep, generalist representation of biological complexity. Although this complexity surpasses straightforward human intuition, advances in artificial intelligence offer a universal framework that leverages data and compute at scale to uncover higher-order patterns6,7. We reasoned that training a model with these capabilities would require data spanning the full spectrum of biological diversity to discover emergent properties similar to those found in other fields8.
We previously demonstrated that machine learning models trained on prokaryotic genomic sequences can model the function of DNA, RNA and proteins, as well as their interactions that create complex molecular machines1,2. Here we present Evo 2, a biological foundation model trained on a representative snapshot of genomes spanning all domains of life. We extend the sequence modelling paradigm to the scale and complexity of eukaryotic genomes through advances in data curation, model architecture, large-scale pre-training, advanced interpretability methods and inference-time prediction and generation approaches.
Emphasizing generalist capabilities over task-specific optimization, Evo 2 represents an important milestone in biological sequence modelling, laying a broad foundation for prediction and design tasks that are relevant to all modalities of the central dogma, that span molecular to genome scale and that generalize across all domains of life.
Evo 2 architecture, training, and data
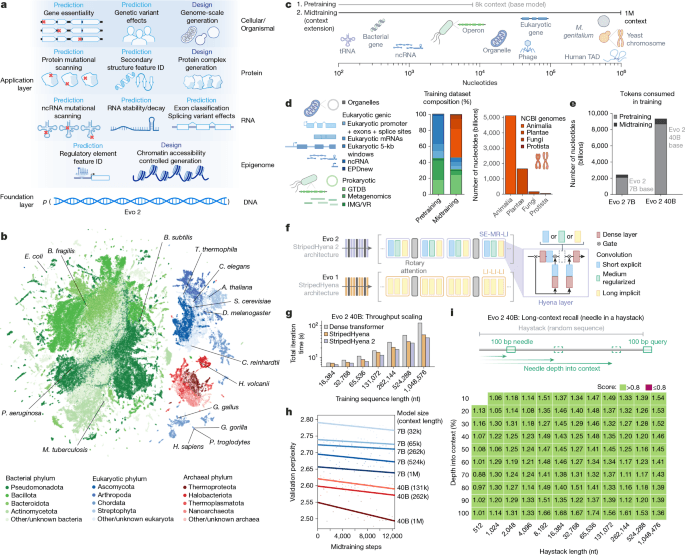
Evo 2 was trained on prokaryotic and eukaryotic genetic sequences, with potential downstream utility for predictive and generative tasks across multiple scales of complexity (Fig. 1a). We trained two versions of Evo 2: a smaller version with 7 billion parameters trained on 2.4 trillion tokens (Evo 2 7B), and a larger version with 40 billion parameters trained on 9.3 trillion tokens (Evo 2 40B). This new training dataset, which we call OpenGenome2, was compiled from curated, non-redundant nucleotide sequence data with a total of more than 8.8 trillion nucleotides from bacteria, archaea, eukarya and bacteriophage (Fig. 1b and Extended Data Fig. 1a).
a, Evo 2 models DNA sequence and enables applications across the central dogma, scaling from molecules to genomes and spanning all domains of life. b, Evo 2 was trained on data encompassing trillions of nucleotide sequences from all domains of life. Each point in the UMAP (uniform manifold approximation and projection) graph represents a single genome in the training dataset that is embedded on the basis of the genome’s k-mer frequencies. Arabidopsis thaliana, Bacillus subtilis, Bacteroides fragilis, Caenorhabditis elegans, Chlamydomonas reinhardtii, D. melanogaster, E. coli, Gallus gallus, Gorilla gorilla, Haloferax volcanii, Homo sapiens, Mycobacterium tuberculosis, Pan troglodytes, Pseudomonas aeruginosa, S. cerevisiae and Tetrahymena thermophila are highlighted. c, A two-phase training strategy was used to optimize model performance while expanding the context length up to 1 million base pairs to capture wide-ranging biological patterns. M. genitalium, Mycoplasma genitalium; TAD, topologically associating domain. d, Novel data augmentation and weighting approaches prioritize functional genetic elements during pretraining and long-sequence composition during midtraining. GTDB, Genome Taxonomy Database; IMG/VR, Integrated Microbial Genomes/Virus database. e, The number of tokens used to train Evo 2 40B and 7B, split into the shorter sequence pretraining and the long context midtraining. f, Schematic of the new multi-hybrid StripedHyena 2 architecture, showing the efficient block layout of short explicit (SE), medium regularized (MR) and long implicit (LI) hyena operators. g, Comparison of iteration time at 1,024 GPU, 40B scale between StripedHyena 2, StripedHyena 1 and Transformers, showing improved throughput. h, Validation perplexity of Evo 2 midtraining comparing the model size and context length, showing benefits with scale and increasing context length. i, A modified needle-in-a-haystack task was used to evaluate long context recall ability up to 1 million sequence length, and shows that Evo 2 performs effective recall at 1 million token context.
Both Evo 2 7B and 40B are trained in two phases to capture biological length scales from molecular to organismal (Fig. 1c–e). Our first stage of pretraining uses a context length of 8,192 tokens, with data weighting focused on genic windows to learn functional genetic elements, followed by a multi-stage midtraining phase over which we extend the context length of Evo 2 to 1 million tokens to learn the relationships between elements across long genomic distances (Fig. 1c–e and Methods). This matches best practice in natural language, in which initial pretraining at shorter context lengths improves both efficiency and overall model quality9,10,11. As in Evo 1, we excluded genomic sequences from viruses that infect eukaryotic hosts from the training data for biosafety purposes. We verified that these data exclusions led to high perplexity on genomic sequences from eukaryotic viruses (Extended Data Fig. 2a), indicating poor language modelling performance in this domain.
Evo 2 uses StripedHyena 2, a convolutional multi-hybrid architecture5 that relies on a combination of three different variants of input-dependent convolution operators12 and attention (Fig. 1f and Extended Data Fig. 1b), improving training efficiency at scale on both short and long sequences, as well as allowing each layer to model interactions at variable distances. StripedHyena 2 provides substantially higher throughput (at 40 billion parameters, up to 3× speedup at 1 million context length) than highly optimized Transformer6 baselines and previous generation hybrid models based on recurrences or long convolutions, such as StripedHyena 1 (ref. 13) (Fig. 1g). StripedHyena 2 also improves loss scaling on DNA against both Transformers and StripedHyena 1 (Extended Data Fig. 1c), thereby achieving both lower prediction error with the same amount of training data and enabling more efficient use of computational resources.
We train up to 1 million base pairs in context length through a multi-stage extension phase, which showed improvements in loss with both model scale and longer context (Fig. 1h). With a synthetic long-context evaluation called ‘needle-in-a-haystack’, we show that Evo 2 can identify and predict the value of a specific 100 base pair sequence (the needle) hidden within 1 million base pairs of random DNA (the haystack), serving as a synthetic quality check that the model can retrieve information from its full context window, as desired for long-context models (Fig. 1i and Extended Data Fig. 1d,e).
By learning the likelihood of sequences across vast evolutionary datasets, biological sequence models capture conserved sequence patterns that often reflect functional importance. These constraints allow the models to perform zero-shot prediction without any task-specific fine-tuning or supervision1,14,15,16. Here, likelihood refers to the probability that the model assigns to a given sequence, where mutations that reduce this probability are predicted to be deleterious. Given that Evo 2 learns a likelihood landscape across all three modalities of the central dogma (DNA, RNA and protein) and all three domains of life, we sought to assess whether Evo 2 could perform mutational effect prediction across these modalities and organisms (Fig. 2a).
a, Evo 2-predicted zero-shot likelihoods can be used to predict the effects of DNA, RNA or protein mutations on molecular function or organismal fitness. WT, wild type. b, Effects on Evo 2 prediction of sequence likelihood caused by mutations along gene start sites for various model species across the domains of life. See Extended Data Fig. 3a,b for additional analyses. T. kodakarensis, Thermococcus kodakarensis. c,d, For different prokaryotic (c) and eukaryotic (d) sequences, the likelihood of different types of mutations in different genomic elements were scored using Evo 2 7B. Scatter represents the median change in likelihood from wild type to mutant sequence per species, coloured by domain (c) or kingdom (d). Horizontal line indicates the median of the scatter distribution. lncRNA, long noncoding RNA; snRNA, small nuclear RNA. e, Mutational likelihoods were used to assess the ability of Evo 2 to differentiate between genomic sequences of model organisms on the basis of their usage of different stop codons. Shown are the standardized median of delta likelihood values across 5 species, where medians were calculated across approximately 4,100 randomly selected mutation loci. M. pneumoniae, Mycoplasma pneumoniae; P. tetraurelia, Paramecium tetraurelia. f, DMS assays were used to assess the Spearman correlation of zero-shot likelihoods from models with experimental assays. Notably, Evo 1 and GenSLM were exclusively trained on prokaryotic datasets. g, Schematic of our single-nucleotide resolution exon classifier based on embeddings from Evo 2. h, Single-nucleotide exon classifiers were trained on embeddings from Evo 2, Nucleotide Transformer (NT) and Evo 1, and were evaluated on the basis of their AUROC across eight held-out species. Performance was compared to SegmentNT-30 kb multispecies (asterisks indicate species in SegmentNT training data), ab initio AUGUSTUS, and to baseline nucleotide content and conservation metrics. D. rerio, Danio rerio; H. vulgare, Hordeum vulgare; S. moell., Selaginella moellendorffii; S. oleracea, Spinacia oleracea; T. cacao, Theobroma cacao; V. vinifera, Vitis vinifera. i, Genome browser track showing predictions from the Evo 2 embedding-based exon classifier scanned across the human STOML2 locus, where the vertical axis is the predicted classifier score and the horizontal axis is genome position. j, Evo 2 predicts genes as essential or nonessential, as determined by experimental gene essentiality assays across bacterial, archaeal and phage species (shown as overlaid scatter) using mutational likelihood of premature stop codon insertions (as a genetic perturbation).
To assess whether Evo 2 captures core biological principles, we first evaluated how single nucleotide variants (SNVs) affect Evo 2 likelihoods in the genomic sequences around the start codons of protein-coding genes. We introduced these mutations at each position in the wild-type sequence and calculated the resulting changes in Evo 2 predicted likelihoods across thousands of such loci (Fig. 2b and Extended Data Fig. 3a). We observed strong changes in the likelihood for mutations within the start codons in both prokaryotes and eukaryotes. This was followed by a three-base periodicity pattern reflecting the triplet codons, with changes at the wobble positions showing lower impact on likelihood. For both prokaryotic and eukaryotic genomes, we observed a pattern upstream of the coding DNA sequence (CDS) that was consistent with the locations of known consensus sequences associated with translation initiation, namely, the Shine–Dalgarno sequence17 for prokaryotes and the Kozak sequence18 for eukaryotes. We also observed similar patterns for SNVs around stop codons (Extended Data Fig. 3b).
Next we measured the effect of mutations across a variety of both noncoding and coding sequences (Fig. 2c,d). Across 20 prokaryotic species and 16 eukaryotic species, we observed changes in model likelihoods consistent with known biological constraints. Non-synonymous mutations, premature stop codons and frameshift mutations caused much larger changes in likelihood than synonymous mutations. In noncoding regions, deletions in transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) had much larger effects than deletions in intergenic and other noncoding loci, reflecting the known essential roles of these RNAs. The 40B model exhibited higher sensitivity to deletions in microRNA (miRNA) and small nucleolar RNA (snoRNA) sequences compared with the 7B model. Evo 2 also predicted that less efficiently translated codons had lower likelihoods than more efficient codons (Extended Data Fig. 3c–e).
Recognizing that our training data contained genomes with distinct genetic codes, we tested how different premature stop codons impacted species that differ in their stop codon usage (Fig. 2e). We found that the model learned the difference between the standard code (stop codons TAA, TAG and TGA), the mycoplasma code (Code 4, stop codons TAA and TAG) and the ciliate code (Code 6, stop codon TGA). When ciliate genomes were artificially recoded to the standard genetic code, Evo 2 predicted mutations from the standard stop codons as deleterious, demonstrating that the model relies on sequence context to determine the appropriate genetic code (Extended Data Fig. 3f).
Although Evo 2 likelihoods reflect the expected importance of different genetic alterations, a key question is whether these likelihoods also correlate with functional effects, which can be empirically measured via deep mutational scanning (DMS) of proteins and noncoding RNAs (ncRNAs). Although state-of-the-art methods for this task tend to leverage both sequence alignments and structural conditioning, general-purpose single-sequence protein language models also learn likelihood distributions that correlate with fitness15. Evo 2 sequence likelihoods correlate with diverse definitions of fitness across nine prokaryotic protein datasets; six eukaryotic protein datasets; and seven datasets of rRNAs, tRNAs and ribozymes (Fig. 2f). Evo 2 is competitive with widely used ProGen language models for protein DMS and with RNA language models for ncRNA DMS, although it underperforms state-of-the-art models on protein DMS. Consistent with observed trends for protein language models, the performance of Evo 2 on these fitness prediction benchmarks begins to saturate and can decrease at the largest model scales19,20,21. We also tested the ability of Evo 2 to predict mutation effects in protein sequences from viruses that infect human hosts. We found no correlation between Evo 2 likelihood and viral protein fitness (Extended Data Fig. 2b), consistent with our data exclusions having the intended effect of weakening both language modelling and downstream performance (Extended Data Fig. 2a). Evo 2 likelihoods also have modest zero-shot association with human mRNA decay rates (Extended Data Fig. 3g and Supplementary Information B.2).
Since Evo 2 learns from eukaryotic genomes, which can be challenging to annotate, we assessed whether its embeddings capture exon–intron architecture. We trained lightweight models on Evo 2 7B base embeddings to develop single-nucleotide resolution classifiers of exon labels (Fig. 2g and Methods). On eight diverse species held out from classifier training, our best classifier achieved areas under the receiver operating characteristic curve (AUROCs) ranging from 0.91 to 0.99 (Fig. 2h,i), outperforming models trained on embeddings from other genomic language models, Nucleotide Transformer22 and Evo 1 (ref. 1), and classification by conservation metrics (local GC content and PhyloP). As a practical baseline, we show that our classifier outperforms ab initio AUGUSTUS23 across all species tested. Evo 2 also outperforms SegmentNT24 on all species outside the SegmentNT training set and on one of the three species in its training set. These results suggest that combining Evo 2 sequence embeddings with supervised approaches can aid the functional annotation of genetic components across diverse species, including non-model organisms.
Beyond molecular or gene-level prediction tasks, we previously showed that high likelihood under Evo 1 is associated with whole organism replication fitness in prokaryotes and phage as quantified by gene essentiality experiments1. Using zero-shot likelihoods to score the effects of premature stop codon insertions into bacterial, archaeal and phage genomes, we found that Evo 2 models performed similarly to Evo 1 and better than other zero-shot methods in predicting gene essentiality across diverse species (Fig. 2j and Extended Data Fig. 3h). On zero-shot prediction of human gene essentiality (Methods), Evo 2 40B (AUROC = 0.66, area under the precision-recall curve (AUPRC) = 0.15) outperformed other genomic language models (AUROC range 0.50–0.59, AUPRC range 0.09–0.12) and performs within the range of four PhyloP conservation scores (AUROC range 0.65–0.71, AUPRC range 0.13–0.21) (Extended Data Fig. 3i), although the overall predictive performance remains modest.
Together, these results demonstrate that Evo 2 captures information across biological modalities and domains of life. Notably, the 7B and 40B models expand predictive capabilities without compromising the prokaryotic insights captured by Evo 1. The utility of both zero-shot likelihoods and simple classifiers trained on Evo 2 embeddings for a variety of predictive tasks across prokaryotic and eukaryotic genomes indicates that Evo 2 provides a strong foundation model for downstream applications in computational biology.
Variant effect prediction represents a critical challenge in genomics, with direct implications for clinical diagnosis and therapeutic development. Genomic language models have previously struggled in eukaryotic variant effect prediction, lagging considerably behind species-specific models that use multiple sequence alignments16,22,25. Evo 2 can perform accurate zero-shot variant effect prediction for both coding and noncoding DNA by considering the changes in the model’s likelihoods after introducing mutations involving single or multiple nucleotides (Fig. 3a).
a, Overview of zero-shot variant effect prediction using Evo 2. Evo 2 was used to assign likelihood scores to human genetic variants, distinguishing pathogenic and benign variants in both coding and noncoding regions. b,c, Zero-shot evaluation of variant pathogenicity within the coding (b; n = 14,319 SNVs, n = 1,236 non-SNVs) and noncoding (c; n = 34,761 SNVs, n = 3,894 non-SNVs) regions. Shown are the AUROCs and AUPRCs for classifying pathogenic and benign variants from ClinVar, across models. For non-SNV evaluations, a modified version of PhyloP was used (Methods). d, Zero-shot evaluation on splice-altering variants in SpliceVarDB, split by exonic (n = 1,181) and intronic (n = 3,769) scoring. e, Evo 2 and other models were used to evaluate BRCA1 variant effect predictions against BRCA1 saturation mutagenesis data, comparing classification of loss-of-function versus functional and intermediate variants in both coding (n = 2,077 SNVs) and noncoding (n = 1,125 SNVs) regions. f, Evo 2 zero-shot likelihood scores plotted for loss-of-function (LOF) versus functional/intermediate variants (n = 3,893), demonstrating the ability of Evo 2 to separate these classes. P value calculated by two-sided Wilcoxon rank sum test. g, Evo 2 embeddings were extracted and concatenated to train a supervised classifier for BRCA1 variant effect prediction. h, Predictions of the supervised classifier on functional/intermediate variants compared with true loss-of-function variants on the test set (n = 789), with decision scores on the horizontal axis. P value calculated by two-sided Wilcoxon rank sum test. i, Comparison of a supervised classifier trained on Evo 2 embeddings on the BRCA1 test set against zero-shot baselines, highlighting the value of using Evo 2 embeddings to build lightweight supervised models.
We used annotations of human clinical and experimentally determined variants to evaluate the ability of Evo 2 to predict biologically important sequence variation. We also contextualize the performance of Evo 2 against a wide range of models, including statistical measures of conservation (for example, PhyloP); unsupervised language models of proteins, RNA and DNA (for example, ESM-1b); supervised splicing prediction models (for example, Pangolin and SpliceAI); and human variant effect prediction models (for example, AlphaMissense, GPN-MSA and CADD).
Using the ClinVar database, we compared the ability of Evo 2 against other methods for predicting the pathogenic effects of human genetic variants across diverse variant classes (Supplementary Data 1). For coding region SNVs, the 40B and 7B models performed competitively, ahead of zero-shot methods, including ESM-2, but behind ESM-1b, GPN-MSA and some PhyloP variants (Fig. 3b). For non-SNV coding variants (for example, insertions and deletions), both Evo 2 models outperformed all other methods; notably, these non-SNV variants are not possible to score by leading models such as AlphaMissense and GPN-MSA (Fig. 3b). For noncoding SNVs, Evo 2 40B ranked first among unsupervised models and only trailed behind supervised models (Fig. 3c). For noncoding non-SNVs, Evo 2 40B outperformed all models tested (Fig. 3c). Across variants stratified by levels of conservation or distance from splice sites, Evo 2 maintains competitive performance among unsupervised models for noncoding variants and the best performance for coding and noncoding non-SNVs out of all methods tested (Extended Data Fig. 4a–c and Supplementary Information B.3).
To further evaluate performance on splice variants, we used SpliceVarDB, a repository containing experimentally validated splicing effects. For both exonic and intronic variants, Evo 2 40B and 7B ranked first among unsupervised models (Fig. 3d). On intronic variants, zero-shot prediction with Evo 2 was competitive with supervised models, slightly trailing SpliceAI and CADD but ahead of Pangolin; on exonic variants, Evo 2 trailed specialized supervised models but outperformed all zero-shot models (Fig. 3d).
We next focused on a dataset measuring functional consequences of variants across both exons and introns of the BRCA1 gene26. Zero-shot prediction with Evo 2 exhibited strong performance on coding SNVs and outperformed all other models on BRCA1 noncoding SNVs (Fig. 3e). Evo 2 7B and 40B achieved better performance than other models when coding and noncoding SNVs were evaluated together, suggesting well-calibrated predictions across included variant types (Extended Data Fig. 5a). When separately considering BRCA1 noncoding variants near or far from splice sites, Evo 2 40B outperformed all tested models, including supervised splicing predictors (Extended Data Fig. 5b). A recently released BRCA2 variant dataset with experimental measurements27 enabled us to extend this analysis to a related gene. Evo 2 surpassed specialized models such as GPN-MSA when predicting coding and noncoding variants together, achieving second-best performance behind CADD, a supervised model (Extended Data Fig. 5c). These results indicate that Evo 2 is an effective zero-shot predictor across diverse types of functional human variants.
Although zero-shot scoring is particularly valuable when task-specific training data are unavailable, model-derived embeddings can also serve as inputs to supervised classifiers that learn task-specific decision boundaries, thereby enhancing both sensitivity and specificity. To illustrate this capability, we assessed whether a simple ridge regression model trained with Evo 2 embeddings exclusively on BRCA1 variants could surpass zero-shot prediction with Evo 2 (Fig. 3g). Given that different layers within large language models capture distinct features, we systematically extracted sequence embeddings from each block of the Evo 2 40B model to identify which layer yielded the most informative features for variant classification (Extended Data Fig. 5d and Methods). Our supervised model achieved a clear separation between loss-of-function variants and all other variants (Fig. 3h and Extended Data Fig. 5e), outperforming zero-shot prediction by Evo 2 40B on the test set (AUROC = 0.95, AUPRC = 0.88) (Fig. 3i). These results underscore how Evo 2 embeddings can be harnessed to train models aimed at more specialized tasks, including those with high clinical relevance.
Unlike the highly constrained sequences typically found in clinical variant datasets which are biased towards coding, splicing or untranslated region (UTR) variants, other regulatory sequences—particularly those distal to genes—exhibit substantially lower conservation. In this context, we used DART-eval to assess how effectively Evo 2 embeddings and likelihoods capture regulatory function28. On zero-shot tasks in DART-eval, Evo 2 40B (chromatin accessibility quantitative trait loci (caQTL) AUROC = 0.58, DNase I sensitivity quantitative trait loci (dsQTL) AUROC = 0.66) outperforms other unsupervised DNA language models, such as Nucleotide Transformer (caQTL AUROC = 0.52, dsQTL AUROC = 0.61), but trails sequence-to-function models trained on accessibility data, such as ChromBPNet (caQTL AUROC = 0.77, dsQTL AUROC = 0.89) (Extended Data Fig. 5f). These results indicate that while multi-species language models trained on sequence alone capture some regulatory information, sequence to function models with task-specific training achieve higher performance in this setting.
On human clinical variant prediction, Evo 2 represents a major improvement over previous multi-species DNA language models across different variant types, with leading performance on non-SNVs (insertions, deletions, duplications), and maintains this performance even in the absence of strong site-independent sequence conservation, although it falls behind supervised models for distal regulatory variants. Furthermore, leveraging the representations in a supervised setting illustrates how Evo 2 embeddings can serve as a foundation for downstream prediction tasks. Notably, Evo 2 is not trained on any human genetic variation or functional genomics data. In sum, these findings support the versatility of Evo 2 as a genome-scale language model for both unsupervised and supervised variant effect prediction.
Evo 2 learns complex representations of genomic sequences without explicit biological labels or annotations. Contrary to the common critique of large language models as black box systems, recent advances in the field known as mechanistic interpretability have demonstrated that sparse autoencoders (SAEs) can reveal latent dimensions that correspond to semantically meaningful features in natural language29,30,31. Without any prior biological annotations or labels, we trained SAEs on Evo 2 representations (or neuron firing patterns), to decompose the model into sparse, high-dimensional representations in which each latent dimension often exhibits human-interpretable patterns (Fig. 4a).
a, SAEs were trained on Evo 2 to extract SAE features associated with interpretable biological function that can be used for annotation, discovery and steering of sequence generations. b, Phage-associated feature activates preferentially on RefSeq-annotated prophages (left and top right) in the E. coli K12 MG1655 genome and fires on phage-derived spacer sequences within CRISPR arrays (bottom right). c, Activations of features associated with ORFs, intergenic loci, tRNAs and rRNAs, in a 100-kb region in E. coli K12 MG1655. d, Activations of features associated with α-helices, β-sheets and tRNAs at an E. coli K12 MG1655 locus containing tufB and a tRNA array ending with thrT (left) and the rpoB–rpoC locus (right). AlphaFold 3 (AF3) structure predictions with feature activations overlaid, of EF–Tu in complex with the tRNA (left) and of RpoB and RpoC in complex (right). e, A feature in the human genome with preferential activation immediately after frameshift mutations over other less deleterious mutation types. f, Features with activation on DNA motifs in the human genome that correspond to transcription factor-binding motifs. g, Features associated with exons, introns and their boundaries in the human genome generalize to a segment of the woolly mammoth genome.
We trained a Batch-TopK SAE32 on Evo 2 representations from layer 26 (Methods). The SAE was trained on representations from 1 billion tokens evenly split across several complete eukaryotic and prokaryotic genomes (Extended Data Fig. 6a–f).
We matched learned SAE latent dimensions, also referred to as features, and known biological concepts by finding features that were enriched in sequence segments containing a particular annotation, a process that we refer to as contrastive feature search (Extended Data Fig. 7a). This revealed diverse features that align with known biological concepts. For example, Evo 2 developed internal representations associated with mobile genetic elements. Feature f/19746 is closely associated with prophage regions across prokaryotes (Extended Data Fig. 7b) and activates on annotated prophages in the Escherichia coli genome, including the cryptic prophage CPZ-55 (Fig. 4b). This feature also activates on spacer sequences within a CRISPR array, which are integrated during CRISPR adaptation from foreign genetic material such as phage DNA (Fig. 4b), as well as after the last CRISPR direct repeat and on synthetic, scrambled spacer sequences, suggesting that Evo 2 associates CRISPR spacers with phage sequences as opposed to directly memorizing phage genomes (Fig. 4b and Extended Data Fig. 7c). This feature also activates on other regions that are not annotated as phage by geNomad33 yet contain genes associated with prophages, such as integrases and invertases (Extended Data Fig. 7d).
Next, we sought to identify concepts associated with canonical biological genomic elements. We identified diverse features corresponding to open reading frames (ORFs), intergenic regions, tRNAs and rRNAs in the E. coli genome (Fig. 4c and Extended Data Fig. 7e,f). We further probed for structural signatures at the protein level and identified features linked to protein secondary structures, such as α-helices and β-sheets (Fig. 4d and Extended Data Fig. 7g,h). These associations highlight the multimodal nature of genome language modelling, capturing higher-order structural information beyond DNA alone.
We extended our analysis to the human genome in search of eukaryotic features. By introducing mutations into thousands of human coding sequences and applying contrastive feature search on a eukaryotic-only SAE, we identified a mutation-sensitive feature (f/24278) that preferentially activates on frameshifts and pre-mature stop mutations (Fig. 4e and Extended Data Fig. 8a,b). We also observed other activations on DNA motifs in the promoter regions of human genes (Fig. 4f, left) that closely resemble the known binding sites of human transcription factors (Fig. 4f, right). Across a random sample of human promoter sequences, Evo 2 unsupervised SAE features have significant hits (q < 0.01, Sandelin–Wasserman similarity) to 70% promoter-enriched motifs (Extended Data Fig. 8f) from the HOCOMOCO v.12 CORE database34 using the TOMTOM motif comparison tool35. For comparison, HOMER36, a specialized motif discovery algorithm, only recalls 35% of the same motifs (Extended Data Fig. 8f). We provide a full report on transcription factor motif-associated features in Supplementary Data 2. These results suggest that Evo 2 contains distinct internal representations of noncoding regulatory elements.
Finally, we identified features that were closely associated with the exon and intron architecture of the human genome, including features that activate preferentially on coding regions (f/15680), introns (f/28339), the first bases of an exon following an intron (f/1050), and the last base of an exon followed by an intron (f/25666) (Extended Data Fig. 8c–e). The coding region feature also activates on bacterial ORFs, suggesting a learned universal representation of coding sequences (Extended Data Fig. 7e,f). Notably, these features demonstrate the ability of the model to learn higher-order sequence dependencies. For instance, exon boundary features (f/1050 and f/25666) integrate signals across splice sites that span multiple nucleotides, and the prophage feature (f/19746) identifies mobile genetic elements requiring kilobase-scale context.
Although we identified these features on the human genome using an SAE trained only on model organisms (including primates, Mus musculus, Xenopus tropicalis and Drosophila melanogaster), we further observed that these features transferred to a genic region within a portion of the woolly mammoth genome37 (Fig. 4g). These results demonstrate that an Evo 2 SAE learns features that transfer across species and suggest utility for genome annotation, although systematic benchmarking against established annotation tools remains necessary.
Overall, we demonstrate that Evo 2 latent representations capture a broad spectrum of biologically relevant signals, from prokaryotic mobile genetic elements and eukaryotic regulatory motifs to protein secondary structure and mutational severity. Since conceptual features for natural language can capture abstract concepts, other Evo 2 SAE features could represent more complex biological patterns (Extended Data Fig. 6f). We have released the SAE models and a visualization tool to facilitate exploration of Evo 2 features for the scientific community.
Beyond its utility on prediction tasks, Evo 2 is also a generative model. We therefore sought to generate DNA sequences from diverse organisms with Evo 2 and assess the quality of designed sequences (Fig. 5a). We previously demonstrated that Evo 1 can respond to DNA prompts to design novel biological sequences2. To evaluate the ability of Evo 2 to respond to genomic prompts, we first assessed performance across six diverse species, spanning archaea, prokaryotes and four eukaryotic lineages (fungi, protists, plants and animals). For each species, we selected highly conserved representative genes and prompted Evo 2 with 1,000 base pairs of upstream sequence plus the first 500–1,000 base pairs of the target gene. We found that Evo 2 achieves gene completion with high amino acid sequence recovery, which improved with scale (Fig. 5b). Evo 2 40B and 7B also demonstrated improved performance over Evo 1 and maintained high sequence recovery throughout long context training (Fig. 5b and Extended Data Fig. 9a).
a, Evo 2 can generate chromosome- and genome-scale DNA sequences using unconstrained autoregressive generation. The model was prompted with portions of the H. sapiens mitochondrial genome, M. genitalium genome and S. cerevisiae chromosome III to generate DNA sequences with similar lengths to those of the native sequences. b, Evo 2 was prompted with both the genomic context and a portion of a highly conserved protein, followed by measuring the sequence recovery of the Evo 2-generated gene completion against the natural gene. c, Predicted rRNA, CDS and tRNA counts in Evo 2-generated mitochondrial sequences using MitoZ compared with the natural H. sapiens mitochondrial genome values. d, Query cover versus sequence identity of generated mitochondrial sequences against nucleotide BLAST hits in the core_nt database with expect threshold of 0.05, coloured by the E-value. e, Visualizations of Evo 2-generated sequences when prompted with a 3-kb sequence from the H. sapiens mitochondrial genome, demonstrating variation that still retains natural synteny patterns of coding sequences. f, AlphaFold 3-predicted structure of multimeric complexes from an Evo 2-generated sequence resembling human mitochondrial DNA. Sequence identity (seq. ID) compares Evo 2-generated proteins with natural proteins found via a BLASTp query. g, Example Evo 2-generated approximately 600-kb DNA sequence. Evo 2 was prompted with the beginning of the M. genitalium genome. Genes are annotated with Prodigal and coloured on the basis of statistically significant sequence similarity to natural proteins (hmmscan E-value < 0.001). h, The fraction of Prodigal-annotated genes with hmmscan hits between Evo 2 40B and M. genitalium generated by Evo 1. i, Distribution of Prodigal-annotated genes from Evo 2-generated M. genitalium compared with the natural genome. j, Distribution of secondary structure from Evo 2-generated proteins compared to natural M. genitalium proteins. k, AlphaFold 3 structure predictions of example proteins found on Evo 2-generated prokaryotic genomic sequences, with high observed structural similarities to natural proteins while diversifying the sequence com-position. l, The native genome sequence from S. cerevisiae chromosome III and an Evo 2-generated DNA sequence of similar length, which was generated by prompting the model with a 10-kb sequence from S. cerevisiae chromosome III, are visualized alongside predicted homologous yeast gene, exon, promoter and tRNA annotations.
Consistent with poor performance for viruses that infect humans on the language modelling task and on function prediction downstreams, Evo 2 also has poor performance on generating proteins from human viruses (Extended Data Fig. 2c). Even when directly trying to elicit a viral protein, Evo 2 had essentially random performance in sequence recovery, preventing Evo 2 from unconstrained or accidental generation of human viral proteins.
To test the ability of Evo 2 to generate DNA at the scale of entire genomes, we assessed its ability to generate all known components of a natural organelle genome. We prompted Evo 2 7B and 40B with portions of human mitochondrial DNA, generating over 250 unique 16-kb sequences (Methods). When annotated with MitoZ38, we found that the generated sequences have the correct number of CDSs, tRNA genes and rRNA genes expected in human mitochondria (Fig. 5c), with varying degrees of sequence similarity to natural genes (Fig. 5d and Supplementary Table 7) while maintaining proper synteny (Fig. 5e). Evo 2-generated mitochondrial sequences contained proteins with predicted multimeric complexes matching those of human mitochondrial proteins (Fig. 5f and Extended Data Fig. 9b,c). The codon usage of generated sequences also closely matched that of the human mitochondrial genome (Extended Data Fig. 9d) and Evo 2 generations on average successfully generated one of each type of expected tRNA, without duplicating the two included in the prompt (Extended Data Fig. 9e).


