
Influenza is an acute respiratory disease that causes 290,000 to 650,000 human deaths each year6. Influenza is caused by an infection with influenza A or B viruses, which circulate in temperate regions as seasonal influenza6. However, rare zoonotic transmissions can cause pandemic influenza outbreaks with high mortality and economic losses7,8. There is current concern that the unexpected susceptibility of dairy cows to avian H5N1 strains may be a path towards a new pandemic9,10,11. Influenza viruses are segmented negative-sense RNA viruses that infect the respiratory tract epithelial cells in humans8. After infection, the eight viral ribonucleoproteins are released into the cytoplasm and imported into the nucleus, where transcription of viral genes into mRNA and replication of the viral genome occur12,13. Each viral ribonucleoprotein contains a genome segment that is encapsidated by multiple copies of the viral nucleoprotein and one copy of the viral RNA-dependent RNA polymerase (FluPol). FluPol consists of subunits PA, PB1 and PB2 and has been structurally characterized2,14,15.
Viral transcripts must contain a 5′ cap structure and a 3′ poly(A) tail to ensure stability, nuclear export and efficient translation16. However, unlike non-segmented negative-sense RNA viruses, the influenza virus genome does not encode enzymes that synthesize a 5′ cap17. Instead, FluPol utilizes capped RNA primers that are cleaved from nascent host transcripts in a process called cap snatching1,5. The FluPol PB2 cap-binding domain binds a nascent 5′ capped host RNA, and the PA endonuclease domain cleaves off 10–15 nucleotides (nt) from the 5′ end. The 3′-terminal nucleotides of this RNA primer then anneal to the 3′ end of the viral genome segment and prime transcription of the viral mRNA14,18,19.
Capped host transcripts are synthesized by cellular Pol II. Pol II transcription starts with assembling a pre-initiation complex consisting of Pol II and the general transcription factors at gene promoters20. To escape from the gene promoter, the largest Pol II subunit RPB1 C-terminal domain (CTD) heptad repeats are phosphorylated at serine 5 and serine 7 by the TFIIH CDK-activating kinase (CAK)21,22. CTD phosphorylation and the growing nascent RNA transcript cause the initiation factors to dissociate from Pol II22,23. Recruitment of the elongation factor DSIF after synthesis of around 20 nt of RNA establishes the early Pol II elongation complex (Pol II–DSIF). This complex is then converted to a paused elongation complex (PEC) containing the negative elongation factor NELF at a transcript length of 25–50 nt (refs. 23,24,25). Synthesis of the 5′ cap occurs co-transcriptionally by the capping enzymes RNGTT, RNMT and CMTR1 (ref. 25) in the context of the Pol II–DSIF elongation complex or the PEC. RNGTT is a bifunctional enzyme that acts as a triphosphatase and guanylyltransferase, creating a GpppN structure at the 5′ end of the Pol II transcript. RNMT and CMTR1 are methyltransferases that add a methyl group to N7 of the cap guanosine and the 2′-OH of the first regular nucleotide, respectively, producing the m7GpppmN cap(1) structure25, which the cap-binding domain of FluPol subunit PB2 tightly binds during cap snatching26,27.
Cap snatching depends on host transcription, as it has been shown that inhibition of Pol II using α-amanitin impairs viral replication3. FluPol localizes primarily at the 5′ end of host genes and associates with the Pol II CTD that is phosphorylated at serine 5 residues, indicating that cap snatching occurs during early phases of Pol II transcription2,28,29,30. Cell-based protein–protein interaction assays indicate that FluPol binds not only to the CTD but also to the Pol II body31. Co-immunoprecipitation–mass spectrometry experiments have shown that the elongation factor DSIF co-purifies with FluPol5,32, and other studies suggest that FluPol depends on the cap(1) structure for cap snatching26. However, how FluPol interacts with the host transcription machinery for cap snatching at the molecular level is unknown.
Here we show that FluPol binds to the transcribing Pol II–DSIF complex for efficient cap snatching. Furthermore, we report two cryogenic electron microscopy (cryo-EM) structures of FluPol bound to a Pol II–DSIF elongation complex before and after endonucleolytic RNA cleavage by FluPol. The structures show that during cap snatching, the PA endonuclease domain of FluPol binds near the RNA exit channel of Pol II and that this interaction is stabilized by DSIF. Furthermore, using cell-based minigenome assays, we confirm that mutation of residues forming the interface between FluPol and the Pol II–DSIF elongation complex reduces FluPol activity in cells. In summary, we present the molecular mechanism of cap snatching by FluPol.
FluPol snatches cap from Pol II elongation complex
To study the molecular basis of cap snatching, we first investigated how the formation of a complex between FluPol and transcribing Pol II (Pol II elongation complex) depends on the cap(1) structure and CTD phosphorylation. We purified Sus scrofa Pol II (99.9% sequence identity to human Pol II, with 4 amino acid differences) from the endogenous source33. Whereas in preliminary studies reconstituting the cap-snatching complex30, we used bat FluPol(H17N10), here we used recombinant, viral promoter-bound FluPol from the influenza strain A/Zhejiang/DTID-ZJU01/2013(H7N9)34,35 (Extended Data Fig. 1a). To reduce RNA cleavage and enhance complex stability, we used the PA(E119D) mutant of FluPol (FluPolE119D; Extended Data Fig. 3j), which has impaired endonuclease activity36,37. A Pol II elongation complex containing a 35-nt cap(1)-RNA, 45-nt template, and non-template DNA was assembled as established previously38. The 35-nt RNA length was chosen considering a 12-nt RNA primer produced by cap snatching19,39, an additional 3 nt bound by the PA endonuclease37, and 20 nt RNA bound within the Pol II elongation complex33.
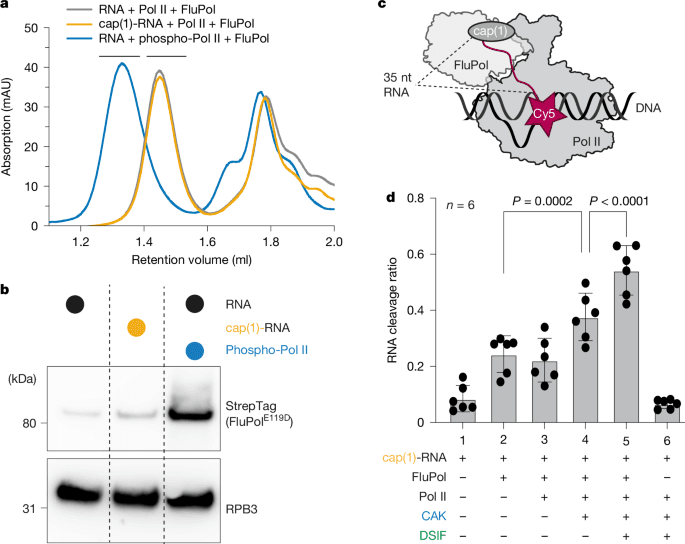
We next monitored binding of FluPolE119D to the Pol II elongation complex by size-exclusion chromatography (SEC) using unmodified RNA and Pol II, cap(1)-RNA, or Pol II that was phosphorylated with CAK. Without CTD phosphorylation and a cap(1) structure, co-elution of FluPol with Pol II could barely be detected (Fig. 1a,b). When a cap(1)-modified RNA was used, the signal for FluPol in the Pol II containing peak slightly increased (Fig. 1b). However, when the Pol II CTD was phosphorylated by CAK, the amount of FluPol associated with Pol II in the peak fractions strongly increased (Fig. 1b). Additionally, the elution volume of the complex peak shifted towards higher molecular weight, indicating the formation of a stable complex (Fig. 1a). Thus, the addition of a cap(1) structure to the RNA has a negligible effect on the interaction between FluPol and the Pol II elongation complex. By contrast, phosphorylation of the Pol II CTD is the main determinant for the recruitment of FluPol to a Pol II elongation complex, consistent with in vivo data demonstrating the importance of the Pol II CTD for viral transcription2,29.
a, Absorbance at 280 nm of analytical SEC runs of Pol II elongation complex containing a 35-nt RNA with or without cap(1) and with or without CAK phosphorylation, and with FluPolE119D. Different colours represent different chromatography runs. Black bars above the chromatogram depict Pol II complex fractions that were analysed by Western blot in b. b, Western blot of Pol II containing peak fractions stained against RPB3 (Pol II) and Twin-StrepTag (FluPol subunit PB2). Individual lanes represent different SEC runs. c, Schematic of the endonuclease cleavage assay. The cap(1)-RNA is Cy5-labelled on the 3′ end. d, The fraction of cleaved RNA (intensity of cleaved product divided by intensity of all bands, see Extended Data Fig. 1c) depends on the factors added. Each point reflects one experimental replicate (n = 6); mean ± s.d. P values were calculated using a linear mixed-effects model (substrate as a fixed effect, experimental replicate as a random effect, two-sided, no multiple testing correction).
We next tested whether the increased affinity of FluPolE119D to Pol II by CTD phosphorylation also results in enhanced endonuclease activity by wild-type FluPol. To monitor RNA cleavage, we developed a fluorescence-based assay using in vitro-capped RNA labelled with Cy5 at the 3′ end (Fig. 1c). We assembled Pol II elongation complexes in vitro essentially as described above, added wild-type FluPol and then visualized the cleaved 3′ end of the RNA that remains attached to Pol II by denaturing PAGE. The primary cleavage product detected was 20–25 nt long, corresponding to the expected 10- to 15-nt primer generated by FluPol (Extended Data Fig. 1b). Additionally, small amounts of an additional cleavage product of around 30 nt were produced. Comparing the different RNA substrates, we did not observe an increase in RNA cleavage by FluPol in the context of a Pol II elongation complex compared to free RNA (Fig. 1d, Extended Data Fig. 1b,c and Supplementary Table 1). However, RNA cleavage increased when we phosphorylated the CTD of Pol II by adding CAK (Fig. 1d and Extended Data Fig. 1b), in line with previous reports40. This suggests that CTD phosphorylation enhances recruitment of FluPol to Pol II and stimulates cleavage of RNA that is bound to Pol II.
Next, we tested whether the presence of the elongation factor DSIF, which binds Pol II during early elongation, stimulates the cleavage of Pol II-bound RNA. Indeed, cleavage of Pol II-bound RNA was stimulated ~1.5-fold when DSIF was added in excess to the Pol II (Fig. 1d and Extended Data Fig. 1b, c). Finally, we tested whether wild-type FluPol can extend the snatched RNA primer using a radioactive FluPol RNA extension assay. We found that FluPol alone can extend the cleaved RNA fragments to some degree. Furthermore, FluPol-dependent RNA extension increases in the presence of a Pol II–DSIF elongation complex, which indicates that the more efficient endonuclease reaction provides more usable RNA primers for FluPol transcription (Extended Data Fig. 1d).
In summary, the cap(1) structure only has a minor effect on FluPol binding to Pol II, whereas CTD phosphorylation by CAK strongly enhances FluPol recruitment and stimulates cleavage of Pol II-bound RNA by FluPol to some extent. Additionally, DSIF, when added to the Pol II elongation complex, significantly enhances RNA cleavage further, suggesting that DSIF is part of the Pol II complex that is recognized by FluPol. Moreover, we have demonstrated that the RNA emerging from the Pol II–DSIF elongation complex can be used to prime RNA synthesis by FluPol. Thus, we conclude that FluPol recognizes the phosphorylated Pol II–DSIF elongation complex as a minimal substrate for efficient cap snatching.
After determining the components required for efficient cap snatching by FluPol in vitro, we next sought to structurally characterize a cap-snatching complex comprising FluPol, Pol II, DSIF and capped RNA by cryo-EM. To that end, we first assembled a Pol II–DSIF elongation complex containing a 35-nt cap(1)-RNA in the presence of the CAK and ATP to phosphorylate the Pol II CTD. To capture the normally transient cap-snatching complex prior to RNA cleavage, we added FluPolE119D at low Mg2+ concentration, a condition in which cleavage is minimal (Extended Data Fig. 1e). The complex was purified and stabilized using GraFix41 prior to cryo-EM sample preparation (Extended Data Fig. 2a). Cryo-EM data acquisition yielded 6,423,874 particles that were further sorted by 3D classification, which yielded a subset of 369,858 particles that show good density for the Pol II–DSIF elongation complex, as well as FluPol resolved at 3.3 Å overall resolution (Extended Data Fig. 2b–h and Extended Data Table 1). From this consensus refinement, we performed focused refinements of FluPol and the Pol II–DSIF elongation complex (with resolutions of 2.90 Å and 2.94 Å, respectively), which enabled us to build and refine an atomic model for the complete cap-snatching complex (Fig. 2a).
a, Two views of the overall structure of the pre-cleavage FluPol–Pol II–DSIF elongation complex complex in cartoon representation except Pol II, which is shown as surface. Dashed black boxes represent the locations of the two interfaces shown in Fig. 3a–c. The structure is shown in a FluPol side view and Pol II top view (top) as well as front view of FluPol and side view of Pol II (bottom). b, The RNA path within FluPol. Proteins are shown as transparent surfaces. The RNA is shown as ribbon tracing of the backbone. Parts of the FluPol model were removed for clarity.
The structure shows that FluPol binds to the Pol II–DSIF elongation complex near the RNA exit channel of Pol II (Fig. 2a). The PA endonuclease of FluPol interacts with the KOWx-4 domain of DSIF that forms a clamp around the exiting RNA in the absence of FluPol33 (Fig. 2a, interface 1). In the complex, KOWx-4 is rotated approximately 180° around its longitudinal axis and shifted by around 22 Å compared with the Pol II–DSIF elongation complex structure33, and the Pol II stalk containing subunits RPB4 and RPB7 is also repositioned (Extended Data Fig. 3a,b). The PB2 cap-binding domain of FluPol inserts between the Pol II subunits RPB1, RPB3 and RPB11 to bind the Pol II dock domain, which is located below the RNA exit channel of Pol II (Fig. 2a, interface 2). In line with our observation that FluPol recruitment to Pol II strongly depends on CTD phosphorylation, we observe density for serine 5 phosphorylated CTD residues in the two previously reported CTD binding sites of FluPol2,42,43 (Extended Data Fig. 3c–f).
We could trace continuous density for most of the RNA from the capped 5′ end in the PB2 cap-binding domain of FluPol all the way to the 3′ end located in the Pol II active site (Fig. 2a,b and Extended Data Fig. 3g,i). This confirms that we successfully resolved the cap-snatching complex prior to endonuclease cleavage. Therefore, we called this structure the pre-cleavage complex. The cap(1) and the first four nucleotides of the RNA are well ordered and tightly bound to the PB2 cap-binding and midlink domains, as observed before18,44. The methylated 2′ OH of the first transcribed base packs against I260 from the PB2 midlink domain (Extended Data Fig. 3h,i), as proposed previously26. The interaction of FluPol with the cap(1) structure is supported by parts of a previously unresolved linker between the KOWx-4 and KOW5 domains of DSIF (SPT5 residues 647–703), which interacts directly with the RNA 5′ end and the cap-binding domain of PB2 (Extended Data Fig. 3h,i). Phosphorylation of serine residues in this linker has been reported to be involved in pause release45. Deleting this linker or alanine mutants of two of the potential phenylalanines that might contact the cap structure only slightly decreases the endonuclease activity of FluPol in vitro (Extended Data Fig. 3k,l).
The nucleotides between the cap-binding domain and the FluPol endonuclease could only be resolved at low resolution (Extended Data Fig. 3g,i), probably owing to the flexibility of this RNA region. This precluded identification of the exact sequence register, although structural modelling (Methods) allows for 9–15 nt of RNA to be placed between the endonuclease and the cap-binding domains (Fig. 2b), in agreement with the primer lengths of 10–15 nt that are produced by co-transcriptional cap snatching in vivo19,39. In summary, we visualized the structure of a pre-cleavage state of FluPol bound to transcribing Pol II during cap snatching, explaining how DSIF stimulates cleavage of Pol II-bound RNA.
Effect of Pol II binding on FluPol in vivo
The biochemical analysis of FluPol endonuclease activity and the structure of the pre-cleavage complex show that FluPol binds the Pol II–DSIF elongation complex, and that the interaction between FluPol and DSIF is important for cap snatching in vitro. Next, we investigated whether the observed interactions between FluPol and the Pol II–DSIF elongation complex are also required for FluPol activity in vivo—that is, in a cellular context. For this purpose, we utilized the pre-cleavage complex structure to identify 16 FluPol residues at the interface with the Pol II–DSIF elongation complex that show high conservation across various influenza strains (Extended Data Fig. 4a,b). We then used a luciferase-based minigenome assay to test FluPol activity in cells after mutating interface residues between PA and DSIF (Fig. 3a, Interface 1), as well as PB2 and Pol II (Fig. 3b,c, Interface 2), to alanines or to their reverse charge counterpart (Supplementary Table 2). As a positive control for defective FluPol transcription, we used the CTD binding mutant PA(K635A)42. Before investigating FluPol activity of these mutants in the minigenome assay, we ensured that all variants are expressed at a similar level as in wild-type FluPol (Extended Data Fig. 5a,b). Of the 26 mutants tested, 19 show a significant reduction in FluPol activity (Fig. 3d and Supplementary Table 3).
a–c, Zoom-ins on the interfaces between the FluPol PA endonuclease domain and the DSIF KOWx-4 domain (a), the FluPol PB2 cap-binding domain and RPB1 (b) or RPB3 and RPB11 (c). Mutated amino acids mutated are shown in stick representation and coloured by heteroatoms if the mutation reduced FluPol activity significantly in a cell-based minigenome assay. d, Cell-based minigenome assay of A/WSN/33 FluPol activity for the indicated PA and PB2 mutants. HEK-293T cells were co-transfected with plasmids encoding PB2, PB1, PA and NP with a model vRNA encoding firefly luciferase. Luminescence was normalized to a transfection control and is represented as percentage of wild-type (WT) FluPol. The K635A mutant was used a transcription-defective control. The dotted line represents background signal as measured in the absence of the PA subunit (∆PA). Each point reflects one biological replicate (n = 3), depicted as mean ± s.d., ***P < 0.001, one-way ANOVA on log-transformed data with Dunnett′s multiple comparisons test referenced to wild-type FluPol.
Mutation of PA residues K104 or E141 to alanine reduced FluPol activity tenfold in the minigenome assay (Fig. 3d). Both residues are close to the conserved SPT5 residue K627 (Extended Data Fig. 4c), with which PA E141 and K104 might form a salt bridge network (Fig. 3a). Individual charge-reversal mutants of K104 and E141 did not further decrease FluPol activity, but combining both alanine mutations did (Fig. 3d). The side chain geometry in this interface is likely to be flexible enough to rearrange in order to compensate for single mutations of the surface charge. Additionally, mutating PA residue Y131, which might be involved in a hydrophobic interaction with SPT5 residue W535, shows the most substantial reduction of luciferase activity of all mutations tested on this surface. PB2 residues D466, T468, S470 and K482 are located at interface 2 between the PB2 cap-binding domain and the RPB1 dock domain (Fig. 3b) and can form hydrogen bonds, as well as salt bridges, with RPB1 residues D400, E404 and R407. From this interface, the T468A and S470A mutants retained around 40% and 20% of wild-type activity, respectively, whereas the other alanine mutations reduced FluPol activity to less than 10% of wild-type activity (Fig. 3d). The charge-reversal mutants D466R and K482E reduce FluPol activity more than the alanine variants. Furthermore, mutation of PB2 residue E452, which is involved in a salt bridge with K17 of RPB11 (Fig. 3c), also reduced FluPol activity in vivo (Fig. 3d). When combined with PB2 D466A, FluPol activity is reduced to a background level. Whereas individual mutations of PB2 R375 or R380—residues involved in the interaction with the RPB3 C-terminal residues H265 and D270—to alanine do not alter FluPol activity, mutating both residues significantly decreases FluPol activity (Fig. 3d). Additionally, the interface residues in RPB1, RPB3 and RPB11 are highly conserved between mammals and birds (Extended Data Fig. 4d–f). These results show that the interface between the PB2 cap-binding domain and the Pol II surface is important for FluPol activity in vivo.
Together, the results show that the integrity of the PA endonuclease interface with DSIF, as well as the interface between PB2 and Pol II, are vital for efficient FluPol activity in vivo. This agrees with our biochemical and structural data showing that the Pol II–DSIF elongation complex is the substrate for cap snatching by FluPol (Fig. 1d).
Mutations at interfaces 1 and 2 reduce FluPol activity in cells; however, this effect could be caused by defects in viral transcription and replication. To determine whether the effects of the mutations are transcription-specific, we performed strand-specific quantitative PCR with reverse transcription (RT–qPCR) in the context of the minigenome assay (Extended Data Fig. 5c and Supplementary Table 4). We then calculated the ratio of influenza mRNA over viral RNA (vRNA) to determine which FluPol mutations specifically affect viral transcription2,46 (Extended Data Fig. 5d). Mutations at the PA–DSIF interface (PA(Y131A) and PA(K104A/E141A)) did not specifically reduce the mRNA/vRNA ratio, suggesting that these residues might be primarily important for replication in vivo. At interface 2, individual alanine mutations of PB2 E452, D466 and K482 did not affect mRNA/vRNA ratios. However, the E452R and D466R charge-reversal mutations, as well as the E452A/D466A double mutation in PB2 led to a reduced mRNA/vRNA ratio (Extended Data Fig. 5d). This shows that PB2 residues E452 and D466 are specifically required for FluPol transcription in vivo. The importance of interface 2 for viral viability was further confirmed by plaque formation assay showing reduced viral titres and plaque diameter for mutants E452R and K482E (Extended Data Fig. 5e, Table 1 and Supplementary Table 5). Viruses expressing PB2(E452A/D466A) could not be rescued by reverse genetics, and viruses with the PB2 D466R mutation acquired a second site mutation R>C (Table 1 and Supplementary Table 6), further demonstrating the importance of this interface for virus viability.
In summary, the PA–DSIF interface mutations do not lead to a transcription-specific FluPol defect in vivo, whereas the PB2–Pol II interface specifically affects FluPol transcription in vivo, highlighting its importance for viral transcription.
DSIF interface is important for endonuclease activity
Both interfaces between FluPol and the Pol II–DSIF elongation complex are required for FluPol activity in vivo, however, we could only find mutations leading to FluPol transcription-specific defects in interface 2. Since a primary replication defect may mask deficiencies in cap snatching and the endonuclease activity of FluPol, we sought to investigate the effect of these interface mutations in vitro. We purified FluPol and DSIF variants with mutations that lead to FluPol activity defects in vivo, and tested them in an endonuclease activity assay using RNA bound to a Pol II–DSIF elongation complex. All FluPol mutants tested here were validated to cleave free RNA with similar efficiency, confirming that the differences observed originate from alterations in interface 1 and 2 (Extended Data Fig. 6c,f and Supplementary Table 1).
Mutations of PA residues Y131, K104 and E141 to alanines lead to a reduction of endonuclease cleavage in the context of a Pol II–DSIF elongation complex (Extended Data Fig. 6a,b). Additionally, when the KOWx-4 domain of DSIF is deleted, endonuclease activity by FluPol is reduced to levels similar to those in a reaction without DSIF (Extended Data Fig. 3k,l). Mutating the potential salt bridge partner in SPT5, K627 (Fig. 3a), to alanine does not significantly reduce FluPol endonuclease activity, whereas an alanine mutation of the SPT5 hydrophobic surface residue W535 leads to reduced RNA cleavage by FluPol. Other interface 1 mutations tested did not alter endonuclease activity (Extended Data Fig. 6b), which is in line with their smaller effect on FluPol activity in vivo (Fig. 3d). This shows that interface 1 is important for efficient endonuclease cleavage. This is additionally confirmed by the reduced plaque diameter of viruses carrying the Y131A mutation or K104/E141A double mutation in PA (Extended Data Fig. 5e and Table 1). Although mutations in PB2 specifically affected FluPol transcription in vivo, these mutations did not alter the endonuclease activity in vitro (Extended Data Fig. 6d,e). This is consistent with the observation that unphosphorylated Pol II alone does not directly stimulate FluPol endonuclease activity (Fig. 1d).
In summary, although PA–DSIF interface mutants do not specifically affect transcription of viral mRNA in vivo, the endonuclease activity of FluPol is reduced for these mutants. This suggests that the stability of the PA–DSIF interface is important for efficient endonuclease cleavage.
The pre-cleavage complex structure reveals the RNA trajectory directly from the cap-binding domain to the endonuclease domain of FluPol. After endonuclease cleavage of the RNA, the newly generated RNA 3′ end must be directed into the FluPol PB1 polymerase active site for RNA extension. It is also unclear whether FluPol stays attached to Pol II after RNA cleavage. To investigate these, we sought to resolve a cap-snatching complex of FluPol bound to the Pol II–DSIF elongation complex under conditions in which the PA endonuclease can cleave the RNA.
To achieve this, we assembled the Pol II–DSIF elongation complex with cap(1)-RNA and FluPolE119D as before, but in the presence of 3 mM Mg2+ (Extended Data Fig. 1e), which led to RNA cleavage during cryo-EM sample preparation (Extended Data Fig. 7a). We then performed cryo-EM as described for the pre-cleavage complex and identified a subset of particles containing cryo-EM density for FluPol and the Pol II–DSIF elongation complex (Fig. 4a and Extended Data Fig. 7b–h). The resulting post-cleavage structure is very similar to the pre-cleavage complex (Extended Data Fig. 8a), except for the path taken by the primer RNA. In particular, we could only trace the RNA from the Pol II active site until the PA endonuclease active site, after which the density discontinues abruptly (Extended Data Fig. 8b–d). This observation suggests that FluPol can stay attached to the Pol II–DSIF elongation complex despite the break in the RNA. The 5′ cap(1) structure remains bound as before in the FluPol cap-binding site. However, the cleaved RNA 3′ end points towards the FluPol polymerase active site, guided by the positive surface charge at the RNA exit channel (PB1 R260, PB2 K214 and R216) (Fig. 4b and Extended Data Fig. 8d,e). The endonuclease cleaves a fragment of 10–15 nt from the Pol II transcript19,39, of which we can observe cryo-EM density for the first 7 nt of the primer in the post-cleavage complex, indicating that the missing nucleotides are disordered. Furthermore, in the post-cleavage complex, the priming loop near the FluPol polymerase active site is still extended and ordered (Fig. 4c and Extended Data Fig. 9a), showing that the snatched RNA primer has not yet base paired with the viral RNA template 3′ end. Thus, FluPol in the post-cleavage cap-snatching state resembles very closely the FluPol pre-initiation complex previously reported18,47 (Extended Data Fig. 9b). Rotation of the cap-binding domain after primer cleavage, therefore, does not seem to be required to direct the primer into the polymerase active site as previously proposed48. Instead, the flexible cap-binding domain is probably fixed in the pre-initiation orientation after binding the Pol II–DSIF elongation complex in the pre-cleavage state (Extended Data Fig. 9c).
a, Overall structure of the post-cleavage FluPol–Pol II–DISF elongation complex in cartoon representation except for Pol II, which is shown as surface. b, Comparison of the RNA path in FluPol between pre and post-cleavage complex. Proteins are shown as transparent surfaces and the RNA is shown as ribbon tracing of the backbone. FluPol polymerase active site Mg2+ atoms are modelled on the basis of the FluPol elongation complex14. Parts of FluPol were removed for clarity. c, Comparison of the FluPol polymerase active site conformations in the post-cleavage (pink) and pre-initiation (Protein Data Bank (PDB): 6RR7, light purple44) states. Only the priming loop, the viral mRNA and the 3′ vRNA are shown.
In summary, RNA cleavage by the FluPol endonuclease results in a new trajectory of the capped primer that is indicative of a FluPol pre-initiation complex. Thus, the PA endonuclease activity on Pol II-bound capped RNA leads to a state of FluPol that is ready to initiate viral transcription with minimal conformational changes.
The results presented here close a major gap in our understanding of the life cycle of one of the most common human viral pathogens. By combining structural, biochemical and cellular approaches, we propose a molecular mechanism of cap snatching by FluPol that involves three major steps (Fig. 5 and Supplementary Video 1). First, FluPol directly binds to the host transcription machinery. The minimal substrate for efficient cap snatching is a Pol II–DSIF elongation complex with a cap(1)-RNA and a phosphorylated Pol II CTD, which is found during early host transcription23,24,33. Second, FluPol endonuclease cleaves the RNA, generating a 10- to 15-nt primer. After cleavage, the new 3′ end of the capped RNA primer is directed towards the FluPol polymerase active site, resulting in a conformation that closely resembles a FluPol pre-initiation complex. Third, the 3′ end of the capped RNA primer can anneal to the vRNA template for viral mRNA synthesis.
Capping enzymes, including CMTR1, synthesize the cap(1) structure on the RNA co-transcriptionally. After capping is finished, CMTR1 dissociates from Pol II. The resulting Pol II–DSIF elongation complex with a capped RNA is a substrate for cap snatching and is bound by FluPol. Then, the FluPol endonuclease cleaves the RNA, and FluPol may dissociate from the Pol II elongation complex surface and initiates viral transcription, whereas Pol II gets terminated.
Building on our mechanistic understanding of co-transcriptional cap snatching, our model also provides insights into how cap snatching is coordinated with host transcription by Pol II (Fig. 5). Several early Pol II transcription states have been structurally characterized, including promoter-proximal pausing49, RNA capping25, pause release into processive elongation (EC*)50 and premature termination51. Comparison of the cap-snatching complex structure with that of the 2′-OH methyltransferase CMTR1 bound to the Pol II–DSIF elongation complex25 shows that binding of FluPol and CMTR1 to Pol II is mutually exclusive (Extended Data Fig. 9e). Since CMTR1 is essential for cap snatching to occur26, it is likely that CMTR1 has to dissociate after the 2′-OH methylation that completes cap(1) synthesis. CMTR1 dissociation from the Pol II–DSIF elongation complex then allows for FluPol binding to the completed cap and the Pol II–DSIF surface, as observed in the pre-cleavage structure (Fig. 2a). NELF binding to the Pol II–DSIF elongation complex establishes the PEC49, which can accommodate FluPol binding without clashes (Extended Data Fig. 9f). However, NELF-E is thought to assist recruitment of the nuclear cap-binding complex to the completed cap52,53. The exact role of the cap-binding complex in cap snatching is not yet understood.
Active elongation in the EC*, in which PAF1c and SPT6 are bound to Pol II, and termination factors such as Integrator or XRN2, however, are sterically incompatible with simultaneous FluPol binding (Extended Data Fig. 9g–i). Thus, the window of opportunity for cap snatching probably opens during Pol II early elongation (Pol II–DSIF elongation complex) and pausing (PEC), and closes after pause release and formation of the EC* or premature termination. Within that window of opportunity, early elongating and paused Pol II represent relatively long-lived substrates for cap snatching as Pol II resides in this phase for several minutes54,55. FluPol binding to the KOWx-4-KOW5 linker of SPT5 might further extend the residence time of the paused Pol II by preventing phosphorylation of the linker, which was shown to be important for pause release45.
The exact fate of FluPol after cap snatching remains unknown. On the basis of our structures, FluPol could remain bound to the Pol II surface during the first steps of viral transcription elongation35 (Extended Data Fig. 9d). However, RNA cleavage probably weakens FluPol binding to Pol II, which seems to correlate with a reduced FluPol occupancy on Pol II in our cryo-EM analysis of the post-cleavage structure. Furthermore, as FluPol transcription progresses, the viral mRNA emerging from the FluPol product exit channel might need more space. These events might lead to FluPol dissociation from the Pol II core, perhaps concomitant with release of the capped RNA from the FluPol cap-binding site and subsequent recruitment of the nuclear cap-binding complex56. Alternatively, termination factors such as Integrator or XRN2 may recognize the FluPol-bound Pol II early elongation complex and could compete with FluPol, triggering its dissociation from Pol II surface (Extended Data Fig. 9h,i), although FluPol could remain bound to the Pol II CTD42. FluPol dissociation and Pol II recycling would allow another round of cellular transcription, leading to a new 5′ cap that can be snatched again by FluPol.
Our structures of co-transcriptional cap-snatching complexes reveal conserved interfaces between FluPol and transcribing Pol II that are crucial for cap snatching. Targeting and disruption of such small protein–protein interfaces by small-molecule inhibitors is inherently difficult57. However, together with recent advances in predicting such interactions58,59, our results may prompt future in silico and experimental studies to identify suitable compounds.
To generate H7N9 FluPol with impaired endonuclease activity, the PA(E119D) mutation was introduced into the PA gene. A pFastBac Dual vector encoding the influenza polymerase heterotrimer subunits of A/Zhejiang/DTID-ZJU01/2013 (H7N9)35, was used as a template for PCR site-directed mutagenesis and Gibson cloning. This method was also used to generate mutated FluPol variants with altered interface with the Pol II elongation complex. Sequencing of all polymerase subunits confirmed the successful introduction of the site-specific mutations in the PA or PB2 gene.
The wild-type FluPol, FluPol PA(E119D) and other FluPol mutants were essentially expressed and purified as described35, with the following modification for all experiments, except the sample preparation for the post-cleavage structure. Initial viruses were generated using transfection in Sf9 cells (obtained from ThermoFisher, not verified in-house), virus propagation in Sf21 cells and protein expression in Hi5 cells (both obtained from Expression Systems, not verified in-house). Instead of ammonium sulfate precipitation as a first step during purification, the supernatant was clarified by ultracentrifugation in a Ti45 rotor (Beckman Coulter) at 45,000 rpm and 4 °C for 1 h.


