
GPCRs comprise a large class of more than 800 receptors in humans that detect a diverse array of stimuli3. Despite their diversity in ligands, each receptor couples to a subset of only four classes of G protein (Gαi/o, Gαs, Gαq/11 and Gα12/13), each with distinct downstream signalling cascades4. GPCR ligands that signal through a reduced subset of endogenous partners (a phenomenon known as biased signalling or functional selectivity) have attracted attention as a means of reducing side-effects and improving the therapeutic windows of drugs5,6. Even with several hundred structures of GPCR–G protein complexes covering most G proteins7, the structural mechanisms by which GPCRs select for their G proteins remain poorly understood8,9, presenting a challenge for the rational design of ligands with bespoke signalling profiles. Recent studies suggest that G protein selectivity is not solely determined by receptor–G protein complementarity in the nucleotide-free state. Rather, evidence points to key roles for transient intermediate states and dynamic interactions involving both G protein conformations1 and receptor motifs, including ICL2 and the transmembrane domain 5 (TM5)–ICL3–TM6 region2.
NTSR1 is a receptor for neurotensin (NTS), a peptide neurotransmitter and hormone that influences diverse physiological processes10. NTSR1 is a promiscuous GPCR, causing significant activation of three G protein classes, with a preference for Gαq/11 > Gαi/o ≫ Gα12/13 (refs. 7,11). Cryo-electron microscopy (cryo-EM) of the nucleotide-free NTSR1–Gi1 complex in detergent and nanodisc revealed a canonical (C) arrangement of the receptor and G protein similar to other family A complexes and an atypical non-canonical (NC) orientation with the receptor rotated by approximately 45° (refs. 12,13). It was proposed that the NC state is an intermediate in the GPCR–G protein association pathway that needs to convert to the C state to allow GTP binding and G protein activation12, although an alternative explanation suggested that the two states reflect the promiscuous G-protein coupling of NTSR1, for example by the NC NTSR1–Gi1 complex, representing a trapped low-energy state that could affect the efficiency of G protein activation and thus selectivity.
Here, we use time-resolved cryo-EM to visualize multiple structures along the GTP-induced G protein activation pathway14,15 for the NTSR1–Gi1 and NTSR1–G11 complexes. Both C and the NC NTSR1–Gi1 complexes are observed bound to GTP, although these are likely to have different allosteric effects on Gα. We resolve high-resolution intermediate states of NTSR1–Gi1-GTP in which contact with Gi1 is maintained largely by interactions between intracellular loop regions and the G protein heterotrimer. By contrast, whereas C and NC conformations occur with nucleotide-free NTSR1–G11, once GTP is added, fewer intermediates are detected that resemble C-like complexes or have the receptor separated from G11. The differences in NTSR1 intermediates with Gi1 and G11, together with dissimilar behaviour for μ-opioid receptor (MOR)–Gi1 activated by GTP14, lead us to hypothesize that sequence divergences in ICL2, ICL3 and the G proteins are drivers of these unique intermediates and cause G11 release from NTSR1 upon introduction of GTP to be substantially faster than Gi1. We demonstrate that these intermediate states are important for rationalizing signalling preferences and subtype selectivity with comparison to previously published assay data, single-molecule fluorescence assays, molecular dynamics simulations and kinetic bioluminescence resonance energy transfer (BRET) data.
Ensembles of apo and GTP-bound NTSR1–Gi1
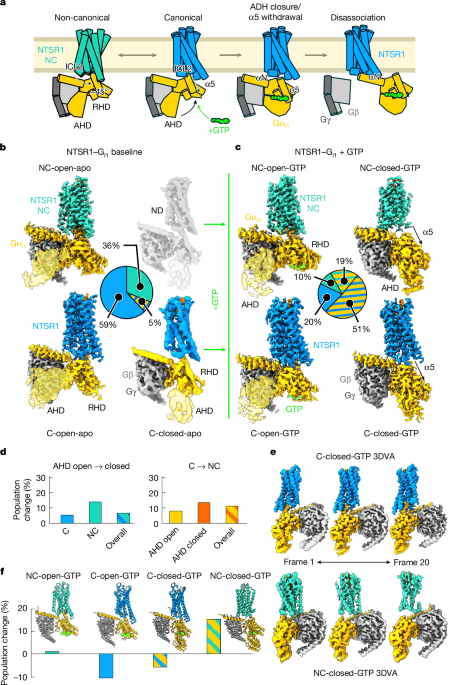
Previous studies have demonstrated that the NTSR1–Gi1 complex possesses C and NC orientations (Fig. 1a) in both detergent and nanodiscs12,13; these complexes either included a Gi1 heterotrimer-stabilizing antibody fragment or were limited to resolutions of around 4.0 Å. To obtain a high-resolution, unbiased reference, we imaged the nucleotide-free complex of Gi1 bound to human NTSR1 in the presence of NTS8–13 peptide, which contains all of the key activating residues that occupy the ligand-binding pocket of NTSR1. We were able to resolve three primary species: the C NTSR1–Gi1 complex with an open alpha-helical domain (AHD) separated from the RAS homology domain (RHD) at 2.1 Å resolution (C-open-apo), the NC complex with an open AHD at 2.3 Å (NC-open-apo), and a third state with the Gi1 AHD in a closed-like conformation with NTSR1 in the C orientation (C-closed-apo) (Fig. 1b and Extended Data Fig. 1a). The ratio of C to NC complexes is roughly 2:1, with around 59% of the particles in the C-open-apo conformation, around 5% in the C-closed-apo conformation and around 36% in the NC-open-apo conformation (Fig. 1b).
a, The previously proposed model for the observed states of the NTSR1–Gi1 complex, in which the NC orientation (turquoise) is an intermediate along the C G protein activation pathway (blue). b, Cryo-EM reconstructions of the nucleotide-free NTSR1–Gi1 complex with wild-type G protein, with their relative populations. Unsharpened maps (translucent golden rod) are overlaid for the AHD to show its positioning. ND, not determined. c, Reconstructions from pooled 6 s and 20 s time points for GTP-induced NTSR1–Gi1 activation, with their relative populations averaged over both time points. d, The percentage change in relative particle populations from the 6 s time point to the 20 s time point; conversion from AHD open to closed (left) and from C to NC (right). e, 3DVA results for the C-closed-GTP and NC-closed-GTP states, showing the most distal frames of the resulting 20-frame videos. f, The percentage change in relative population from 6 s to 20 s for all four states, overlaid with a proposed model for the transition between the observed intermediate states along the GTP-induced activation pathway.
The high resolution of our nucleotide-free Gi1 maps provide an opportunity to more precisely compare the C and NC states. The major conclusions about the conformational changes in the two receptor states remain, with the primary difference lying in the TM7–helix 8 (H8) region, with an unusual and more inactive-like conformation of the NPXXY motif for the NC state (Extended Data Fig. 2a). On the basis of the cryo-EM maps, ICL2 becomes more dynamic in the NC state, probably as a result of looser packing with the G protein (Extended Data Fig. 2b). There are also subtle differences in the G protein, with the β6–α5 (TCAT) loop being more disordered in the C state (Extended Data Fig. 2c). The different ICL2 positions drive changes in the β2–β3 loop (Extended Data Fig. 2c,d), probably resulting in the α1 helix being slightly more extended in the C state (Extended Data Fig. 2c). Three-dimensional variability analysis (3DVA16) identifies little further heterogeneity in key activation motifs outside of transient ordering of the linker I region (Extended Data Fig. 2e). We could not detect any AHD closed particles with an NC receptor orientation, possibly owing to the differing allosteric effects of the receptor ICL2 region on the β2–β3 loop, α1 helix and the α5 helix (Extended Data Fig. 2c). To explore these effects, we performed molecular dynamics simulations starting from the open and closed AHD C and NC states, which demonstrated greater AHD dynamics in both open and closed states for the NC orientation (Extended Data Fig. 2f), although without sampling full open–closed transitions in the timescales used, we cannot rule out other explanations for the greater AHD opening with the NC orientation receptor.
We then performed time-resolved cryo-EM experiments probing the GTP-induced activation of the NTSR1–Gi1 complex14,15, producing grids at 6 s and 20 s after addition of GTP. Processing the 6 s and 20 s data separately revealed four shared intermediate states (Extended Data Fig. 1d), and pooling all of the data for both time points14,15 enabled us to reconstruct each state at 2.4–3.0 Å resolution. The first two states correspond to open AHD conformations with the receptor in either C or NC orientations with GTP present (C-open-GTP and NC-open-GTP), revealing that the NC conformation is capable of binding GTP (Fig. 1c). We additionally resolve two states with the AHD closed against the RHD, the α5 helix dropped to engage with the nucleotide base, and the receptor remaining in an active-like conformation (Fig. 1c and Extended Data Fig. 3a). In the first of these states, the receptor occupies an orientation similar to the C state but with the α5 helix withdrawn (C-closed-GTP) (Fig. 1c and Extended Data Fig. 3b). The second state more closely resembles the NC state, although with further rotation along the membrane axis to almost 90° from the C state (NC-closed-GTP) (Fig. 1c and Extended Data Fig. 3c).
In both AHD closed states, retraction of the α5 helix leaves ICL2 and ICL3 responsible for an increased proportion of the ordered contacts between receptor and G protein (Extended Data Fig. 3d,e). This is particularly prominent in the NC states, in which ICL2 accounts for 75% of contacts in NC-closed-GTP compared with only 6% of contacts in the NC-open-apo structure (Extended Data Fig. 3d,e). In the NC-closed-GTP state, the ICL2 contacts involve F17434.51, T17834.55 and R1824.39 (in Ballesteros–Weinstein notation17), Gβ and the Gα αN helix (Extended Data Fig. 3f). The TM5–ICL3–TM6 region of the NC-closed-GTP state is also poised with TM6 between the β2–β3 loop and the α5 helix, with ICL3 partially disordered but filtering between the β2–β3 loop and the αN helix (Extended Data Fig. 3g). In both C-closed-GTP and NC-closed-GTP states, despite the α5 helix occupying the retracted conformation, the tip of α5 is still observed inside an intracellular cavity of the receptor, although the first principal component of 3DVA reveals that, whereas nearly all of the C-closed-GTP particles have some degree of α5 tip engagement and a stable receptor (the two extreme states from this 3DVA denoted C-closed-GTP and C-closed*-GTP), the NC-closed-GTP particle stack contains particles in which greater α5 disengagement is correlated with a poorly resolved receptor (Fig. 1e, Extended Data Figs. 3a,h and Supplementary Videos 1 and 2). The reduced α5 helix engagement and propensity for receptor dissociation in the NC-closed-GTP state suggests that this state may occur after the C-closed-GTP state—that is, further along the pathway of total complex dissociation. To ensure that these results were not an artefact of the detergent environment, we repeated the NTSR1–Gi1 6 s time point in a MSP1D1 nanodisc, enabling identification of all four states in the two-dimensional class averages (Extended Data Fig. 4a), with a slight enrichment of particles in C conformations relative to those in NC conformations. Further, we were able to obtain sub-3.0 Å reconstructions of the C-open-GTP and C-closed-GTP states, demonstrating nearly identical conformations to detergent (Extended Data Fig. 4b).
In the pooled time-resolved cryo-EM data, addition of GTP substantially shifts the conformational equilibrium compared with the nucleotide-free baseline, with AHD closed conformations increasing from around 7% of particles to the predominant species at around 70% regardless of whether the receptor is C or NC (Fig. 1b,c). The ratio of C to NC remains roughly 2:1 for both AHD open and AHD closed states (Fig. 1c). By comparing the fraction of particles in each state at 6 s and 20 s, we can track the evolution of the system over time, with the first trend being the expected shift towards particles in AHD closed states as time progresses (Fig. 1d). Notably, we also observed a time-dependent increase in the relative populations of particles in the NC-like states, particularly NC-closed-GTP (Fig. 1d). This observation, together with the differing allosteric effects on AHD closure and the 3DVA result demonstrating an unstable receptor in the NC-closed-GTP particle stack, supports a model similar to the one originally proposed, in which the NC state represents an intermediate that both precedes the C state during nucleotide exchange and follows the C state during GTP-induced complex dissociation. However, given that the NC state is still capable of binding guanine nucleotide, we cannot rule out direct NC G protein activation without a C state. Intermediate C and NC NTSR1–Gi1 states are sufficiently stable to resolve using time-resolved cryo-EM but are driven by ICL–Gi heterotrimer interactions, raising the question of to what extent this ensemble of states occurs with the other G proteins and how they influence those signalling pathways.
To our knowledge, there are no cryo-EM structures of the nucleotide-free wild-type NTSR1–G11 complex, and few such structures exist for any receptor. Structure determination of Gq-coupling receptors has almost exclusively used miniGq18, which lacks the AHD and has several other modifications19. We purified sufficient nucleotide-free NTSR1–G11 complex to obtain cryo-EM datasets, revealing similar structures to those that we observed for NTSR1–Gi1 (Fig. 2a), although with some differences. Both C and NC orientations of the receptor exist with NTSR1–G11, indicating that the NC conformation is not specific to Gi. In the C orientation, we observe two major conformations of the AHD, each with substantial populations, corresponding to a relatively typical AHD open state (C-open-apo) and a second state in which the AHD is partly closed (C-P-closed-apo) (Fig. 2a). The difference between G proteins is likely to stem from differences in the α1 helix-linker I region between Gi and G11. Whereas NTSR1–Gi1 exhibited varying degrees of α1 helix extension (Extended Data Fig. 2c), α1 is fully or almost fully extended in all of the states observed for G11 (Extended Data Fig. 5a). The linker I region of G11 contains a glycine immediately following the histidine at the end of α1 (Extended Data Fig. 5b), conferring greater flexibility to the linkage between RHD and AHD. Kinetic BRET assays examining Gβγ release for NTSR1–Gi1 and NTSR1–Gq reveal that when the linker I regions are swapped between Gi and Gq, the kinetics of Gi1 signalling are significantly impaired, whereas the similar swap onto Gq marginally increased kinetics, suggestive that Gi1 signalling is optimized for a sharp open–closed transition (Extended Data Fig. 5c). Complexes with the C orientation still outnumber those with the NC orientation, although with a slightly diminished ratio of around 1.5:1. Even though the relative population is higher for both the NC-open-apo state and a state with some degree of closure of the AHD compared with our results for Gi1, we still do not observe any appreciable population of a NC state with any degree of AHD closure.
a, Nucleotide-free baseline cryo-EM maps of the NTSR1–G11 complex, with their relative populations. b, The two states observed in time-resolved cryo-EM of the NTSR1–G11 complex separated with three-dimensional classification. Insets display an overlay of the maps and models for the changes in the switch II (Sw II) and switch III (Sw III) regions. c, Comparison of the NTSR1–Gi1 and NTSR1–G11 C-closed-GTP state, highlighting the rotation of the receptor and slight repositioning of ICL2. d, Alignment of the structure of G11 with the NTSR1 NC-closed-GTP complex on the αN helix and RHD, highlighting likely unfavourable interactions in a hypothetical NTSR1–G11 NC-closed-GTP complex. e, Biotinylated Tris-Ni2+-NTA-immobilized, nucleotide-free complexes of NTSR1 bound to either Gi1 or G11 were subjected to pre-steady state addition (at t = 1 s) of imaging buffer with or without 1 mM GTP. The loss of labelled NTSR1 molecules from surface-immobilized G protein complexes were fit with either one (for buffer only) or three (for buffer with GTP) exponential functions. Plotted data represent mean (lines) and s.d. (shaded areas) from two independent repeats.
Although the presence of C and NC complexes was shared by Gi1 and G11, profound differences were observed in time-resolved cryo-EM studies. In contrast to what we observed for NTSR1–Gi1, we did not identify any population of particles in an NC-like orientation or with an open AHD after addition of GTP. Rather, in the sole high-resolution heterogeneous reconstruction result, the receptor occupies a C orientation with closed AHD and retracted α5 helix. 3DVA and three-dimensional classification of this state reveals further heterogeneity that can be grouped primarily into two states (Fig. 2b and Supplementary Video 3). One state has substantial overlap with the C-closed-GTP state of NTSR1–Gi1. In the other, the α5 helix is completely disengaged from the receptor, which becomes highly dynamic, and further changes are observed in G11 itself. In the G11 heterotrimer, the switch II region begins to move away from Gβγ and towards the switch III region, which also moves upward to engage switch II (denoted C-closed*-GTP) (Fig. 2b and Extended Data Fig. 6a). Examining the 3DVA results for the first principal component of the Gi1-C-closed-GTP particle stack shows that this state also undergoes switch II and switch III region dynamics, although with less significant rearrangement than G11-C-closed*-GTP. In Gi1 the reordering is primarily restricted to the base of switch II, with R208, R205, Q204 and G203 shifting towards Gα to interact with E245, switch III and GTP (Extended Data Fig. 6a–c). However, these changes in Gi1 occur without the accompanying changes in the upper switch II residues, including W211, that are observed with G11. Further, the overall movement of Gα away from Gβγ is reduced in Gi1-C-closed-GTP compared with G11-C-closed-GTP (Extended Data Fig. 6a). Rearrangements in the switch II–switch III region are likely to be some of the final changes in Gα before full GTP-induced activation and complex dissociation, and several of the rearranging residues highlighted have been demonstrated biochemically to be key for GTP-induced activation (including the GRE motif G203/G208, R208/R213, E245/E250 (ref. 20) and the key catalytic and oncogenic Q204/Q209 (ref. 21)). Therefore, we anticipate that even subtle differences in the behaviour of these important microswitches between Gi1 and G11 could be key drivers of different activation kinetics.
The orientation of NTSR1 differs slightly between the C-closed-GTP states of Gi1 and G11, with subtle effects on G protein engagement. ICL2 still engages the cleft between the β2–β3 loop and the α5 helix of G11, with F17434.51 inserted into a hydrophobic region, but several key residues in the cleft are substituted (noticeably, the β2–β3 loop K192 of Gi1 is swapped for E197 of G11). ICL2 (and thus the entirety of NTSR1) is rotated by approximately 10° in G11-C-closed-GTP compared with Gi1-C-closed-GTP (Fig. 2c), which is necessary to avoid a steric clash and/or unfavourable electrostatic interactions with the αN R37 of G11 (corresponding to A31 of Gi1; Extended Data Fig. 6d). The percentage of contacts between NTSR1 ICL2 and G11 again increases substantially from the C-open-apo to the C-closed-GTP state, from 21% to 44%, suggesting a role for ICL2 remains in G11 interaction (Extended Data Fig. 6e,f).
We aligned the Gα11 subunit to the NC-closed-GTP state observed with Gi1 to explore why such an intermediate is absent in the presence of GTP for G11 (Fig. 2d). One potential roadblock is a lysine residue (K33 of Gα11; G27 of Gαi1) which would extend towards the ICL2 lysine K17734.54 of the receptor in the NC orientation (Fig. 2d), generating an unfavourable electrostatic interaction. The αN–β2–β3 loop–α5 region that TM5–ICL3–TM6 threads through in this orientation also bears substantial differences in sequence between G proteins (Fig. 2d and Extended Data Fig. 6g), although it is difficult to pinpoint unfavourable residues without a resolved structure. More broadly, comparing sequences in intermediate-state receptor-binding G protein regions reveals substantially greater sequence variation between major subtype families compared with members of the same subtype, highlighting the potential for these regions to more generally drive the stability of GPCR–G protein intermediate states and coupling selectivity (Extended Data Fig. 6h), which is supported by previous bioinformatics and FRET (Förster resonance energy transfer) work22,23.
Given the comparative lack of stable intermediate states with G11 compared to Gi1 in the GTP activation pathway, it would seem likely that activated G11 release from NTSR1 is significantly faster than Gi1, and that this is an important component of the higher levels of G11 signalling compared with Gi1 observed in assays. To probe this, we performed pre-steady-state single-molecule fluorescence-based experiments of GTP-induced NTSR1–G protein complex activation. In brief, a nucleotide-free complex of fluorescently labelled NTSR1 and either Gi1 or G11 was tethered to a microfluidic device (Fig. 2e and Methods). At low illumination intensity, GTP induced the dissociation of NTSR1–Gi1 and NTSR1–G11 complexes with time constants of around 37 s and 15 s, respectively, approximately 2.5 times slower for Gi1 release compared with G11 (Fig. 2e and Supplementary Table 1). These findings corroborate time-resolved cryo-EM analysis in which NTSR1–Gi1 complexes undergo GTP-induced dissociation through more stable intermediate conformations than NTSR1–G11 complexes.
Four NTSR1 residues that are thought to be involved in interactions with Gi1 in the NC, but not C, nucleotide-free complex (S93 and L94 in ICL1, R2946.26 at the base of TM6, and H3738.52 in H8) were previously demonstrated with mutagenesis to be able to selectively reduce Gi1 signalling compared with Gq12. Given that both C and NC states exist for the NTSR1–G11 nucleotide-free complex, it is worth considering how this G protein selectivity is being conveyed. The interaction between R2946.26 and the α5 helix occurs in both Gi1 and G11 NC nucleotide-free G protein complexes (Extended Data Fig. 7a), whereas ICL1 is largely dynamic in both the G11-NC-open-apo and Gi1-NC-open-apo state maps (Extended Data Fig. 7b). There is no interaction between G11 and the receptor H8 H3738.52 in the G11-NC-open-apo state (Extended Data Fig. 7a); this interaction is not well-resolved in our Gi1-NC-open-apo structure (Extended Data Fig. 7c) and the interaction is long for a strong hydrogen bond in the previously reported Protein Data Bank (PDB) structure 6OSA12. Thus it would seem unlikely these mutations selectively knock out the NC-open-apo state of Gi1 and not that of G11. Some of the selectivity of these mutations is better rationalized by considering the intermediate states; for example, R2946.26 of NTSR1 in the Gi1-C-closed-GTP state is pointing towards the retracted α5 helix and can be modelled into residual map features forming a salt bridge with D350 on the α5 helix (Extended Data Fig. 7d,f). By contrast, owing to the rotation of NTSR1 in the G11-C-closed-GTP state compared with the Gi1-C-closed-GTP state, R2946.26 is moved almost 5 Å away from the α5 helix and is thus unlikely to be interacting with the G11 E353 that is equivalent to Gi1 D350 (Extended Data Fig. 7e). It is worth noting that for muscarinic acetylcholine receptors (mAChRs), interactions between TM5–TM6 and the α5 helix alone can differentiate between Gi- and Gq-coupled mAChRs24. The α5 helix sequence, including the terminal hook, having a role in G protein subtype selectivity is an even more broadly observed phenomenon25,26,27. By capturing the AHD closed intermediates, we gain additional insights into how the α5 helix hook samples the intracellular GPCR cavity when the α5 helix is in the retracted position. In contrast to the nucleotide-free state, the cryo-EM maps suggest that ICL1 may be more involved in interacting with Gi1 in the C GTP-bound intermediate than the NC (Extended Data Fig. 7g). Thus, our time-resolved cryo-EM data reveal that these mutations probably do not uniquely discriminate between C- and NC-like states, but can help rationalize their effects on Gi1 versus G11 signalling.
To explore how other GPCR–G protein activation intermediates may influence G protein selectivity, we compared our NTSR1 results to previous work that examined GTP-induced activation of the MOR–Gi1 complex14, which reveals similarities and differences in the intermediate states observed and their apparent stabilities (Fig. 3a and Extended Data Fig. 6a). Two high-population intermediate states exist for NTSR1–Gi1 with a closed AHD, withdrawn α5 helix and active-conformation receptor (Fig. 3b). By contrast, only a small fraction of the MOR–Gi1 particles (when bound to an equivalently strong agonist such as NTS8–13)28,29 are resolved with both a closed AHD and active receptor (G-ACT-1; Fig. 3a), and only with minimal α5 helix withdrawal and incomplete AHD closure (Extended Data Fig. 8a). Rather, full, and particularly superagonist-bound, MOR–Gi1 has almost complete disengagement of the receptor from the G protein once the AHD fully closes (Fig. 3a and Extended Data Fig. 8a), with a pseudo-stable MOR–Gi1 AHD closed complex only occurring with partial agonists that have collapsed back to the inactive receptor conformation (Extended Data Fig. 8b).
a, Populations and structures for the intermediate states observed in the GTP-induced activation of the MOR–Gi1–lofentanil complex. b, Populations and structures for the intermediate states observed in the GTP-induced activation of the NTSR1–Gi1 complex. c, Sequences of the NTSR1 and MOR ICL2 region and alignment of the active-state receptor structure of MOR (green) with the C-closed-GTP state of NTSR1, highlighting ICL2 differences. d, Alignment of the active-state receptor structure of MOR with the NC-closed-GTP state of NTSR1, highlighting the C-closed-GTP state of NTSR1 and differences in ICL2. e, Alignment of the active-state receptor structure of MOR with the NC-closed-GTP state of NTSR1, highlighting the C-closed-GTP state of NTSR1 and differences in TM5–ICL3–TM6, together with the sequence of the TM5 and TM6 regions of NTSR1 and MOR.
By aligning the active structure of MOR to the Gi1-C-closed-GTP and Gi1-NC-closed-GTP structures for NTSR1 (Fig. 3c,d), we can identify several key residue changes mainly centred on ICL2 that would destabilize similar intermediates with MOR. Substitution of the ICL2 F17434.51 of NTSR1 for V17534.51 would reduce hydrophobic packing in both states (Fig. 3c,d), and for the C-closed-GTP state would probably modulate the allosteric effect on the α1–β2–β3 loop–α5 region (Fig. 3c). This may contribute to intrinsic differences in AHD opening between NTSR1–Gi1 and MOR–Gi1, as NTSR1–Gi1 possesses a higher propensity for AHD closure14 (Fig. 3a and Extended Data Fig. 8c). D17934.55 of the MOR ICL2 (T17834.55 in NTSR1) would be positioned near D193 of Gi1 in a hypothetical Gi1-C-closed-GTP intermediate state, producing unfavourable charge–charge repulsion (Fig. 3c). Finally, a Gi1-NC-closed-GTP state with MOR would be incapable of forming the interaction between the TM5–ICL3–TM6 of the receptor and the αN–β2–β3 loop–α5 of Gi1 given the substantially shorter TM5 of MOR (Fig. 3e). Molecular dynamics simulations of the Gi1-NC-closed-GTP state performed with both NTSR1 and with the receptor replaced by MOR revealed that the Gi1-NC-closed-GTP NTSR1 state is stable over multiple 2 μs trajectories, but a hypothetical Gi1-NC-closed-GTP MOR complex undergoes rotation of the receptor back towards a more C-like arrangement (Fig. 4a). Simulating the Gi1-C-closed-GTP complex again reveals that with NTSR1 the complex is stable over 2 μs, as is the MOR–Gi1-C-closed-GTP complex, although MOR has on average 3–4 Å longer ICL2–β2–β3 loop distances, despite the smaller sidechain of V17534.51 compared to the equivalent F17434.51 (Extended Data Fig. 8e). In summary, two relatively stable intermediate complexes with Gi1 are observed with active NTSR1 but not MOR, probably because of differences in ICL2.
a, Molecular dynamics simulation results from n = 5 independent runs for the NTSR1–Gi1 NC-closed-GTP structure (turquoise traces) and the equivalent complex with MOR (green traces). The distances between ICL2 and the β2–β3 loop (left) and between ICL3 and the α4 helix (right) are used as a metric for whether the complex resembles the C-closed-GTP or NC-closed-GTP state. b, Cartoon of the kinetic bioluminescence resonance energy transfer (BRET) assay design. c, Kinetic BRET traces for the NTSR1 and MOR constructs tested in this work. WT, wild type. d, Heat maps of kinetic BRET assay results examining Gi and Gq signalling for NTSR1 and MOR with ICL2 and/or ICL3 swapped. Experiments were performed with three (NTSR1) or four (MOR) independent experiments with four technical replicates each. e, Cartoon of the ICL2 point mutation locations and heat maps of kinetic BRET assay results examining Gi and Gq signalling for NTSR1 ICL2 point mutants. Experiments were performed with three (NTSR1) or four (MOR) independent experiments with four technical replicates each.
It has been reported for several family A GPCRs that ICL2 and ICL3 sequences contribute substantially to G protein subtype selectivity30,31,32,33,34, which together with of our structural results highlight these regions as areas for study with mutagenesis. We generated ICL2 and ICL3 chimeric constructs between NTSR1 and MOR for testing in BRET assays of agonist-induced Gβγ release (Fig. 4b). Given our time-resolved cryo-EM results, we hypothesize that swapping ICL2 and the TM5–ICL3–TM6 region of NTSR1 with MOR would inhibit the ability of NTSR1 to form C-closed-GTP and NC-closed-GTP states. This should produce an outsized effect on signalling, despite both receptors canonically coupling strongly to Gi and the nucleotide-free structures suggesting relatively small effects of ICL swaps (Extended Data Fig. 8d). Consistent with this hypothesis, maximum Gβγ release was significantly degraded with NTSR1MOR-ICL2 compared with wild-type NTSR1 (Fig. 4b), and the ICL2 swaps slowed the kinetics of Gi–Gβγ release for both receptors by around 50% (Fig. 4c,d). The effect of NTSR1MOR-ICL2 on Gq maximum Gβγ release was similar to results for Gi (approximately 50% reduction in both cases); however, kinetics of Gq signalling were substantially more affected, with an approximately 82% reduction. By contrast, the NTSR1MOR-ICL3 swap had less of an effect on Gi–Gβγ release kinetics than NTSR1MOR-ICL2, although there was a similar effect on maximum activation, and was even less impactful on Gq signalling (Fig. 4c,d). These effects are consistent with a major role for ICL2 contact-driven intermediate states in signalling. Notably, swapping both ICLs of NTSR1 with those of MOR (NTSRMOR-ICL2/3) almost completely knock out Gq signalling while retaining some level of Gβγ release for Gi (Fig. 4c,d). However, MOR with both NTSR1 ICLs (MORNTSR1-ICL2/3) did not induce Gq signalling (Extended Data Fig. 8f), highlighting that other regions, including the TM7–H8 interface, have a role in signalling, and contrasting with recent work demonstrating that the introduction of ICL2 from a GPCR that couples only to Gq is sufficient to introduce some Gq activation to MOR33. Together with our analysis of the quadruple mutants that selectively knock out NTSR1–Gi1 signalling, these results highlight that the nucleotide-free state alone does not capture many salient features for G protein selectivity.
We also tested point mutations of several of NTSR1 ICL2 residues (F17434.51L/V, F17434.51V, K17734.54L and T17834.55D) either to the corresponding residue from MOR or other common residues from family A GPCRs (Fig. 4e). F17434.51L and F17434.51V had little negative effect on the maximal signalling through Gq, and only F17434.51V slowed Gq kinetics, whereas maximal signalling was impaired at Gi1 with F17434.51V, consistent with the proposed importance for the position 34.51 hydrophobic packing observed in the Gi1 closed intermediate states. The K17734.54L mutation substantially impaired the maximal signalling of both G proteins, but sped up Gi1 signalling kinetics, whereas T17834.55D mildly impaired kinetics and maximal signalling for both G proteins. Analysis of ICL2 sequences for receptors that couple exclusively to Gi/o or Gq/11, or primarily couple to both (Extended Data Fig. 8g), shows sequence biases consistent with the assay results and structural data. Gi-exclusive receptors have a strong tendency towards valine in position 34.51 (32% of receptors) and absence of phenylalanine (2%, a single receptor), whereas no Gq or Gi/Gq receptors have a valine at 34.51, with phenylalanine present in 10% of Gq receptors and 25% of Gi/Gq receptors (it has also been reported that for many Gs-coupled receptors, Y or F is required in this position34). The 34.54 position also has a strong bias towards a cationic residue in Gi/Gq receptors (58%) and Gq receptors (30%) but not in Gi receptors (11%). This agrees with prior work with a primary Gi-coupled receptor (muscarinic receptor M2R), where valine at 34.51 was dispensable for signalling and could be mutated to alanine with minimal effect, but where a bulky hydrophobic 34.51 residue was more important for secondary Gi coupling35. These results provide a more granular view of how ICL2 influences G protein subtype selectivity, although a more complete picture will probably require additional visualization of the G protein association pathway and/or assays that uniquely probe association and dissociation separately.
Here we use time-resolved cryo-EM to reveal several distinct intermediate states for GTP-induced activation of the NTSR1–G protein complex for members of two different families of G proteins. These results shed light on the role of the NC orientation of NTSR1–G protein complexes and demonstrate that they are capable of binding to GTP. We further demonstrate that GTP-induced release of G11 occurs much more rapidly compared with Gi1, with far fewer resolvable intermediate states. This significant rate difference in the release of GTP-bound G protein is likely to be a major contributing factor to the more efficient activation of Gq/11 G proteins by NTSR1 compared with Gi/o7,11, emphasizing the importance of the full cycle of association–nucleotide exchange–dissociation in signalling kinetics.
Canonically important regions for G protein subtype selectivity of both GPCRs (ICL2 and ICL3 (refs. 2,24,30,31,32,33,34)) and G proteins (α5 and β2–β3 loops22,23,24,25,26,27) are shown to interact to stabilize unique intermediate-state structures of NTSR1 with Gi1 and G11. This in turn enables us to rationalize aspects of G protein subtype selectivity that are not apparent from equilibrium structures of nucleotide-free complexes. Sequence conservation in key regions including ICL2 suggests that although strong trends occur in both promiscuous versus selective receptors and primary versus secondary coupling35, there are no unique amino acid compositions in singular motifs that can be used to trivially predict signalling. This raises the likelihood that although other receptors presumably share similar GTP-bound G protein intermediate states to MOR or NTSR1, there may be further novel intermediates that are yet to be resolved, particularly for receptors that have non-conservative substitutions in positions essential for the observed intermediate states (for example, a cationic R or K instead of F/V34.51). Comparison of the sequences of G protein regions that interact with the receptor revealed substantially less variability between members of the same family than across families, consistent with previous bioinformatics and signalling work on the importance of these regions22. Nonetheless, we hypothesize that even small sequence differences in G proteins could affect the relative population of intermediate states (C versus NC in the case of NTSR1) and contribute to signalling differences observed for receptors across members of the same G protein family, and may mediate the signalling bias observed with some compounds36.
These results suggest several future avenues for building on the present work to allow a more complete understanding of NTSR1 signalling and G protein subtype selection. Full characterization of NTSR1–G protein coupling will require visualization with time-resolved cryo-EM of NTSR1 interacting with its GDP-bound G protein partners to form complex and release nucleotide, as there are likely to be substantial similarities but also subtle differences in the association and dissociation pathways. Building on these structures, biochemical and biophysical methods for probing the conformational ensemble of GPCR–G protein complexes, including double electron–electron resonance (DEER)37 and single-molecule FRET38, together with approaches such as the single-molecule fluorescence assays leveraged here, can probe the kinetics of each step of the signalling process and measure the lifetimes of specific intermediates, resolving questions of which states correspond to rate-limiting steps. Together with molecular dynamics simulations39 and machine learning methods, this can enable the determination of full free energy landscapes for signalling and show how the energy landscape is perturbed by different G protein signalling partners and different ligands bound to the receptor. These insights will provide the necessary structures and dynamic information to guide the design of GPCR ligands with precise functional selectivity profiles, allosteric modulators and intracellular ‘glue’ compounds40,41,42,43,44, and other next-generation molecules that will significantly facilitate the production of improved GPCR drugs.
Expression and purification of G protein heterotrimer
G protein heterotrimer was expressed as described14 in Tni cells (Expression Systems; isolated from Trichoplusia ni; no validation or mycoplasma testing) with viruses for Gα, Gβγ and Ric-8a and snap frozen in liquid nitrogen for later use. To purify wild-type G protein, cell pellets were thawed and resuspended in a lysis buffer containing 20 mM HEPES pH 7.5, 1 mM EDTA, 5% glycerol, 1 mM MgCl2, 5 mM β-mercaptoethanol, 100 µM GDP, Pierce Universal Nuclease, and protease inhibitor cocktail, and gently stirred for 30 min at 4 °C. The lysate was then subjected to ultracentrifugation at 100,000g for 35 min, and the supernatant was discarded. Pellets were resuspended in solubilization buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 1% sodium cholate, 5% glycerol, 1 mM MgCl2, 5 mM β-mercaptoethanol, 100 µM GDP, Pierce Universal Nuclease, and protease inhibitor cocktail, and allowed to gently stir for 1 h at 4 °C prior to ultracentrifugation at 100,000g for 35 min. Solubilized protein was supplemented with 30 mM imidazole and incubated for 1 h with Ni-NTA beads. The beads were collected by centrifugation at 300g, packed into a column, and washed with 10 column volumes of a series of buffers containing 30 mM imidazole and 50% solubilization/50% E2 buffer, 25% solubilization/75% E2 buffer, 12.5% solubilization/87.5% E2 buffer, and 100% E2 buffer (E2 buffer contained 20 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, 1 mM MgCl2, 5 mM β-mercaptoethanol, 100 µM GDP, and 0.05% lauryl maltose neopentyl glycol (LMNG)/0.005% cholesterol hemisuccinate (CHS)). Protein was eluted with 3 column volumes of buffer containing E2 buffer and 250 mM imidazole, and was incubated with 1 mg of 3C protease per 50 mg of protein with overnight dialysis at 4 °C in dialysis tubing. The next day, the Ni-NTA column was washed with 10 column volumes of buffer containing E2 and 30 mM imidazole, and overnight cleavage was loaded over the nickel and flow through was collected. The column was washed with an additional 2 column volumes of the E2 buffer and 30 mM imidazole, and flow through was collected. Protein was concentrated subjected to size-exclusion chromatography with a Superdex 200 column and a buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, 1 mM MgCl2, 100 µM TCEP ((Tris(2-carboxyethyl)phosphine)), 20 µM GDP, and 0.01% LMNG/0.001% CHS. Fractions were pooled, concentrated, and snap frozen in liquid nitrogen for later usage. For single-molecule experiments instead of cutting the His tag and performing reverse chromatography, the purified G protein heterotrimers were then mixed with biotinylated Tris-NTA (Sigma, supplemented with equimolar NiCl2) at a molar ratio of 2:3 and incubated on ice for 1 h. The G protein heterotrimer and biotinylated Tris-Ni2+-NTA complexes were subsequently purified using size-exclusion chromatography (Superdex200, Cytiva).
Pellets containing NTSR1 were expressed in Sf9 cells (Expression Systems, no validation or mycoplasma testing) with the baculovirus system as described12 and snap frozen in liquid nitrogen for storage. To purify NTSR1, cell pellets were thawed and lysed in hypotonic lysis buffer containing 10 mM HEPES pH 7.5, 1 mM benzamidine, Pierce Universal Nuclease, protease inhibitor cocktail, 1 mM EDTA, 1 mM MgCl2, and 100 µM TCEP, and gently stirred for 1 h at 4 °C. Subsequently, membranes were collected by ultracentrifugation at 100,000g, the supernatant was discarded, and pellets containing the membranes were resuspended in solubilization buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl, 1 mM benzamidine, Pierce Universal Nuclease, 1 mM MgCl2, 100 µM TCEP, and protease inhibitor cocktail. Detergent was then added dropwise while gently stirring at 4 °C to a final concentration of 1% LMNG/0.1% CHS/0.1% sodium cholate. After 3 h of stirring, insoluble debris was removed via ultracentrifugation at 100,000g, supplemented with 20 mM imidazole, and loaded over a TALON resin column. The columns were washed with 10 column volumes of buffer containing 20 mM HEPES pH 7.5, 20 mM imidazole, 500 mM NaCl, and 0.1% LMNG/0.01% CHS. Protein was eluted with 2 column volumes of buffer containing 20 mM HEPES pH 7.5, 250 mM imidazole, 250 mM NaCl, 10% glycerol, and 0.01% LMNG/0.001% CHS. Purified receptor was concentrated and subjected to size-exclusion chromatography with a Superdex 200 column and a buffer containing 100 mM NaCl, 20 mM HEPES pH 7.5, 5% glycerol, and 0.01% LMNG/0.001% CHS. Peak fractions were pooled, concentrated, and further supplemented with 5% glycerol, then snap frozen for use for complexation.
Formation and purification of NTSR1–G protein complex
Complex formation was initiated by mixing NTS8–13-bound receptor (NTSR1 incubated with NTS8–13 for 1 h) with a ~1.25 molar excess of G protein and incubating for 1 h on ice before addition of apyrase to remove GDP and HRV 3C protease to cleave the GFP tag from NTSR1 and incubation on ice overnight. Complex was then diluted in a buffer containing 100 mM NaCl, 20 mM HEPES pH 7.5, 10 μM NTS8–13, 0.01% LMNG/0.001% CHS, 5 mM CaCl2 and loaded onto M1 Flag resin, washed with a buffer of 100 mM NaCl, 20 mM HEPES pH 7.5, 10 μM NTS8–13, 0.005% LMNG/0.0005% CHS, 5 mM CaCl2 to remove excess G protein. NTSR1–G protein complex, and then eluted with a buffer of 100 mM NaCl, 20 mM HEPES pH 7.5, 10 μM NTS8–13, 0.005% LMNG/0.0005% CHS, 200 μg ml−1 Flag peptide and 1 mM EDTA. Eluted complex was concentrated and loaded onto a Superdex 200 size-exclusion column in a buffer of 100 mM NaCl, 20 mM HEPES pH 7.5, 10 μM NTS8–13, 0.001% LMNG/0.00033% GDN/0.0001% CHS, 2 mM MgCl2. Fractions of complex were concentrated to 6–10 mg ml−1 for cryo-EM studies.
Preparation of NTSR1–G protein complex in nanodisc
NTSR1–Gi1 complex was formed overnight as described in detergent, and then incubated for 1 h with solution containing MSP1D1 belt protein (Sigma) mixed with 3:2 POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine):POPG (1-palmitoyl-2-oleoylphosphatidylglycerol) at a 1:5:400 ratio of complex:MSP1D1:lipid. Two 1 h incubations with Bio-Beads SM-2 were performed followed by a final 8 h incubation with fresh Bio-Beads SM-2. NTSR1–G protein complex was then further purified as described for the detergent complex with Flag and size-exclusion chromatography, but in the absence of detergent. Nanodisc formation was confirmed with SDS–PAGE and concentrated to 2–4 mg ml−1 for cryo-EM studies.
Cryo-EM sample preparation and data collection
UltrAufoil R1.2/1.3 300 mesh grids were glow discharged in a Pelco unit for 45 s at 10 mA before being used for grid freezing. 3 μl of sample was pipetted onto a grid in a vitrobot held at 100% humidity and 4 °C, 0.3 μl of 10 mM GTP was added for the time-resolved cryo-EM grids, and the grids were blotted and plunge frozen in liquid ethane with a preprogramed blot and wait time to achieve 6 s and 20 s time points, chosen based upon prior studies with β2AR and MOR and consistent with the lifetimes observed in single-molecule based fluorescence assays. Detergent data were collected on a G4 Titan Krios equipped with a K3 direct electron detector and a Bioquantum energy filter. Details of each data collection can be found in Extended Data Tables 1 and 2 and Supplementary Table 2, but in brief, grids were imaged with a nominal defocus range from −0.6 to −1.6 μm and a target dose of 55 e− Å−2 over 40 total frames in super-resolution mode. Nanodisc data were collected on a G3 Titan Krios equipped with a Falcion IV/Selectris energy filter, with similar parameters to the detergent datasets.
All data were processed in cryoSPARC45. Raw videos were aligned with patch motion correction and contrast transfer function (CTF) estimation was performed with patch CTF estimation. Particle picking was performed with template-based picking using templates for GPCR–G protein complexes. Extracted particles were subjected to two-dimensional classification to remove noise particles. Further cleaning was performed with iterative rounds of ab initio model generation with multiple classes and heterogeneous refinement. Once particle stacks were largely free of junk particles, non-uniform refinement was applied to obtain reconstructions. 3DVA was run on these reconstructions to probe for further heterogeneity, and the first principal component analysed. Final reconstructions came from non-uniform refinement, in cases of particularly high-resolution maps CTF refinement and reference-based motion correction were performed. In the case of the Gi1 NC-open-apo and Gi NC-open-GTP states, manual sharpening and preferred orientation correction, respectively, were applied to improve the map quality. Particle counts for comparing results were based upon the pre-orientation filtering numbers. A graphic example of the processing for the major experiments performed in this work is provided in Extended Data Fig. 1. Consistent with prior time-resolved cryo-EM studies of GPCR–G protein complexes, data from individual time points was merged to boost the resolution of reconstructions, and assessment of the time point-related changes in populations were based on whether particles contributing to final reconstructions came form 6 s or 20 s micrographs, although analysis of the individual time point data separately provided similar results (Extended Data Fig. 1d).
The prior cryo-EM structures of NTSR1–Gi1 were docked for all initial complex structures (PDB: 6OSA and 6OS9)12, while the Gi AHD closed state was docked from the prior MOR–Gi1 time-resolved work (PDB: 9ODL). Initial structures for G11 were obtained from crystal structures of the G11 heterotrimer (PDB: 8QEH)46. Manual model building was performed in Coot47 with refinement in Phenix48. Details of cryo-EM map and model refinement are found in Extended Data Tables 1 and 2. Euler angle distribution plots, FSC curves, local resolution plots, and selected map–model agreement panels are provided in Extended Data Figs. 7 and 8 and Supplementary Figs. 1 and 2.
GPCR–G protein complexes were prepared by extending the N-terminus of Gα, the C-terminus of the receptor, and the C-terminus of Gγ to add enough residues to include their sites of lipidation (if not already modelled) by using residual low-resolution map features for that respective state. The PPM webserver47 was used to orient systems which were then solvated in a box of POPC/CHS at a concentration of ~5% CHS (chosen to provide the presence of this important signalling molecule without significantly perturbing the bulk bilayer properties), TIP3P water49, 100 mM NaCl, and 15 mM MgCl2 using the CHARMM-GUI50. Simulations were performed in NAMD51 with the CHARMM36 forcefield52,53,54 using a Langevin thermostat and Nose-Hoover Langevin piston barostat at 1 atm with a period of 150 fs and decay of 75 fs. Periodic boundary conditions were used with nonbonded interaction smoothing at 10 Å to 12 Å with long-range interactions handled with particle mesh Ewald. A 2 fs timestep was employed with SHAKE and SETTLE algorithms used. All non-hydrogen, non-water/ion atoms were restrained with harmonic restraints of 1 kcal mol−1 Å−2 and the systems were minimized for 1,500 steps before gradual heating from 0 to 303.15 K in 20 K increments with 0.4 ns of simulation per increment, with an additional 10 ns of equilibration at 303.15 K. A further 10 ns of equilibration with harmonic restraints only applied to non-hydrogen protein atoms were performed, followed by another 10 ns of equilibration with harmonic restraints only applied to Cα atoms. The first 30 ns of unrestrained simulation was discarded from averaging as equilibration, with the following simulation considered production. All simulations were run with 5 replicates with different initial seeds for random assignment of velocities. VMD55 and Python scripting were employed for analysis. Distances for ICL2–β2–β3 were calculated from the centre of mass of residue F174 for NTSR1 and V175 for MOR and K192 of Gi1, while TM6-α4 distances were calculated based upon residue 6.26 in Ballesteros–Weinstein notation and residue D309 of Gi1. AHD–RHD distances were calculated based on the centre of mass of the two domains.
HEK 293 cells (ATCC, revalidated by STR and mycoplasma negative) were transfected in 6-well plates with plasmids encoding a GPCR (NTSR1, MOR or variants), Gα subunit (Gαi1 or Gαq), Venus1-155Gγ2, Venus155-239Gβ1 and memGRKct-Nluc at a ratio of 1:0.8:0.4:0.4:0.1 μg per well using linear PEI. For activation of Gq heterotrimers PTX-S1 (0.2 μg) was also transfected to prevent activation of endogenous Gi/o heterotrimers. For activation of Gi/o heterotrimers 1 μM YM-254890 was added to prevent activation of endogenous Gq heterotrimers. Kinetic BRET assays were performed 24 h after transfection using a BMG Lumistar plate reader equipped for simultaneous measurement of donor and acceptor emission. Cells were resuspended in DPBS containing 5 μM furimazine, and NTS8–13 (1 μM final concentration) or DAMGO (10 μM) were injected after collecting a 10 s baseline. NTSR1 assays were performed in triplicate with technical quadruplicates while MOR assays were performed in quadruplicate with technical quadruplicates. ICL2 swaps exchanged the nine residues from 34.51 to 4.39 while ICL3 swaps exchanged the residues from 5.68 to 6.25.
Purified wild-type NTSR1 was mixed with iodoacetamide-derivatized LD555 (custom synthesis by Lumidyne technologies) at a molar ratio of 1:0.3 and incubated at room temperature for 1 h. The labelling reaction was quenched by L-cysteine (10 mM). The excess dye was removed by purifying the labelled NTSR1 on a G50 desalting column (2 ml).


